伴异源性横纹肌肉瘤的卵巢支持-间质细胞瘤:常见DICER1突变,TERT启动子、TP53与BRAF突变可能参与肉瘤转化
时间:2026-05-09 21:25:25 热度:37.1℃ 作者:网络
深度解析医学证据,lxfs.net为你支撑决策
伴异源性横纹肌肉瘤(RMS)的卵巢支持-间质细胞瘤(SLCT)极为罕见。尽管 SLCT 中异源性成分的存在高度提示存在 DICER1 突变,但这类肿瘤(包括伴异源性 RMS 的 SLCT)的分子变异在很大程度上仍不明确。本研究旨在描述这类罕见肿瘤的临床病理特征,并对部分病例进行详细分子分析,探究潜在的频发性及成分特异性基因变异。
本文报道 11 例伴异源性 RMS(结蛋白与肌细胞生成素阳性)的卵巢 SLCT,其中 10 例符合胚胎型 RMS,1 例为多形性 RMS。患者年龄呈双峰分布:7 例(64%)年龄≤33 岁(平均 20 岁),4 例(36%)年龄≥52 岁(平均 60 岁)。所有肿瘤均为单侧。除 RMS 成分外,11 例中 8 例(73%)含其他异源性成分,包括胃肠型黏液上皮(5 例)和未成熟软骨(3 例)。11 例中 7 例(64%)行二代测序(NGS)分析。所有分子检测的肿瘤(7/7,100%)均携带 DICER1 热点突变。其中 6 例(86%)同时存在第 2 个无义或移码功能缺失型 DICER1 突变。1 例仅存在 p.D1810Y 热点突变,肿瘤由高级别肉瘤伴局灶性横纹肌母细胞分化(结蛋白与肌细胞生成素局灶阳性)构成,符合多形性 RMS;多形性肉瘤成分同时呈现突变型 p53 表达。除 DICER1 突变外,所有病例均存在 TERT 启动子(c.-124C>T,4 例)或 TP53 突变(3 例),且二者互斥。对 2 例进行成分特异性分析显示,SLCT 与 RMS 成分共享相同的 DICER1 热点突变,支持单克隆起源。其中 1 例 TERT 启动子(c.-124C>T)体细胞突变仅见于 RMS 成分;另 1 例 TERT 启动子突变见于两种成分,而 BRAF p.V600E 突变仅见于 RMS 成分。
本研究表明,大多数伴异源性 RMS 的 SLCT(86%)存在 DICER1 双突变(1 个热点突变 + 1 个无义/移码功能缺失突变),支持现有关于 DICER1 突变与 RMS 异源性成分相关的认知,出现此类成分应启动基因咨询。本研究结果还提示,除 DICER1 外,TERT 启动子与 TP53 突变可能参与特定成分的肿瘤转化。
研究背景
卵巢支持 - 间质细胞瘤(SLCTs)是罕见的性索 - 间质肿瘤,占原发性卵巢肿瘤不足 0.5%。在现行世界卫生组织(WHO)分类中,其分为高分化、中分化、低分化及网状亚型。高分化肿瘤通常呈良性生物学行为,中分化、低分化及网状亚型则可能表现为恶性。高分化肿瘤多为单纯型,而中分化、低分化及网状亚型常混合存在,构成形态学谱系。形态学上,这类肿瘤可呈现多种组织学表现,包括异源性成分。分子层面,SLCT 可分为三个亚型:DICER1 突变型(患者年龄小、雄激素症状、中 / 低分化、网状或异源性成分)、FOXL2 c.402C>G 突变型(绝经后患者、雌激素症状、中 / 低分化、无网状或异源性成分)及 DICER1/FOXL2 野生型(患者年龄中等、无网状或异源性成分,包括所有高分化肿瘤)。然而,FOXL2 突变组的分类存在一定争议,因其临床与分子特征与成人颗粒细胞瘤(FOXL2 突变、绝经后、雌激素症状、无异源性及网状成分)存在重叠。尽管文献报道不一,平均超过半数的 SLCT 存在 DICER1 基因 RNase Ⅲb 结构域体细胞热点突变,其中高达 69% 为胚系突变。
约 20% 的 SLCT 含异源性成分,最常见为良性胃肠型黏液上皮。约 5% 的病例存在异源性间叶成分,典型为软骨或骨骼肌,后者通常表现为横纹肌肉瘤(RMS)。伴异源性 RMS 成分的 SLCT 极为罕见,可发生于儿童与成人患者,总体预后较差。尽管 SLCT 中网状与异源性成分的存在高度提示 DICER1 突变,但伴异源性 RMS 成分的 SLCT 的分子变异仍缺乏深入研究。本文报道 11 例伴异源性 RMS 成分的卵巢 SLCT,描述其临床病理特征,并对 7 例进行详细分子分析,揭示频发性及有时成分特异性基因变异。
研究结果
临床特征:
11 例患者临床特征总结于表 1。3 例有详细个人史与家族史(病例 2、10 为会诊病例,病例 5 为院内病例),均无 DICER1 相关肿瘤的记录;其余病例均为会诊病例,无详细病史资料。患者年龄 7–71 岁,平均 34 岁,中位 27 岁。值得注意的是,年龄呈双峰分布:7 例(64%)≤33 岁(平均 20 岁,中位 20 岁),4 例(36%)≥52 岁(平均 60 岁,中位 58 岁)。所有肿瘤均为单侧,均以盆腔包块为首发表现。4 例血清 AFP 升高,多数病例术前未检测血清 AFP。病例 7 为 27 岁孕妇,孕 10 周,表现为左侧卵巢肿块破裂伴腹腔积血。病例 10 表现为多毛症,发现左侧卵巢包块,睾酮与 AFP 升高,卵巢切除后两项标志物均恢复正常。肿瘤大小 16–28cm(平均 22cm,中位 23cm),右侧卵巢 7 例,左侧卵巢 4 例。诊断时 2 例存在卵巢外播散:病例 4 累及大网膜,病例 10 肿瘤侵犯肠系膜、盆腔侧壁与前腹壁。3 例有随访资料,均接受化疗。病例 5 卵巢切除后 27 个月死于转移性疾病;病例 6、10 分别随访 12、44 个月,无病生存。
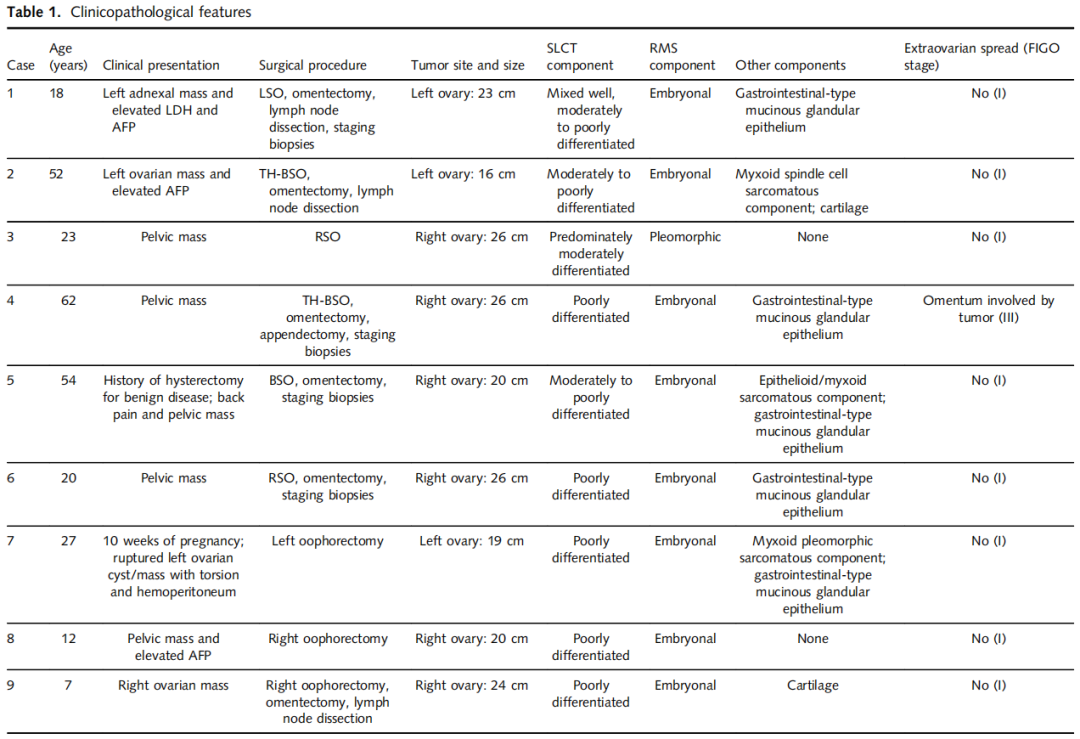

表1
伴异源性 RMS 成分的 SLCT 病理与免疫组化:
10 例(病例 1、2、4~11)呈现典型 SLCT 伴异源性胚胎型 RMS 特征。所有病例 SLCT 成分为中至低分化,形态多样,包括小管(闭合型、空心型)(病例1,图 1A、B)、弥漫片状(病例1,图 1C)、岛状 / 条索状排列(病例2,图 1D)、微囊性结构伴较大扩张囊腔含嗜酸性分泌物类似甲状腺滤泡(病例4,图 1F、图 1G)、网状结构(病例7和11,图 1H)、缎带样或长条索状(病例8,图 1I)。所有病例均存在间质细胞成分,偶见稀疏分布。免疫组化显示,SLCT 成分 SF-1(10 例检测均阳性)(图 1E)、钙视网膜蛋白(6 例检测均阳性)、抑制素(10 例检测均阳性)阳性,详细结果见表 2。
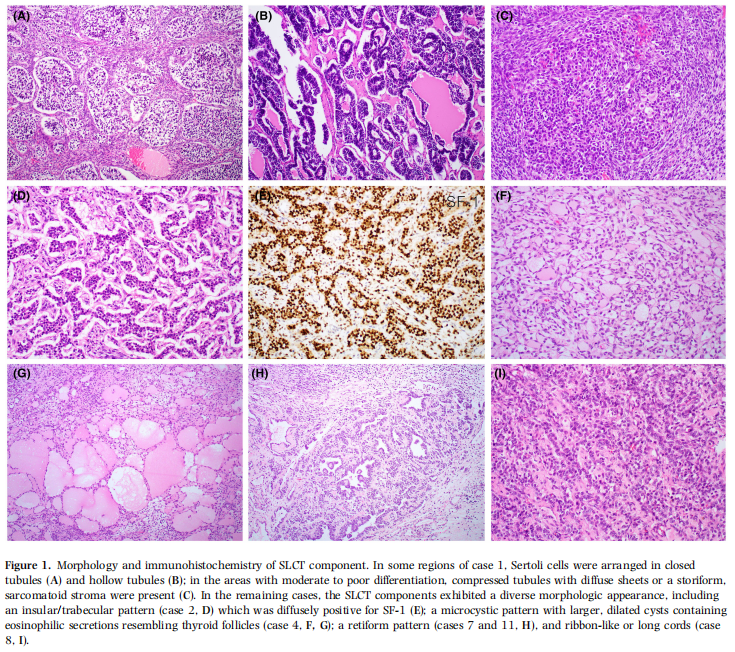
图1
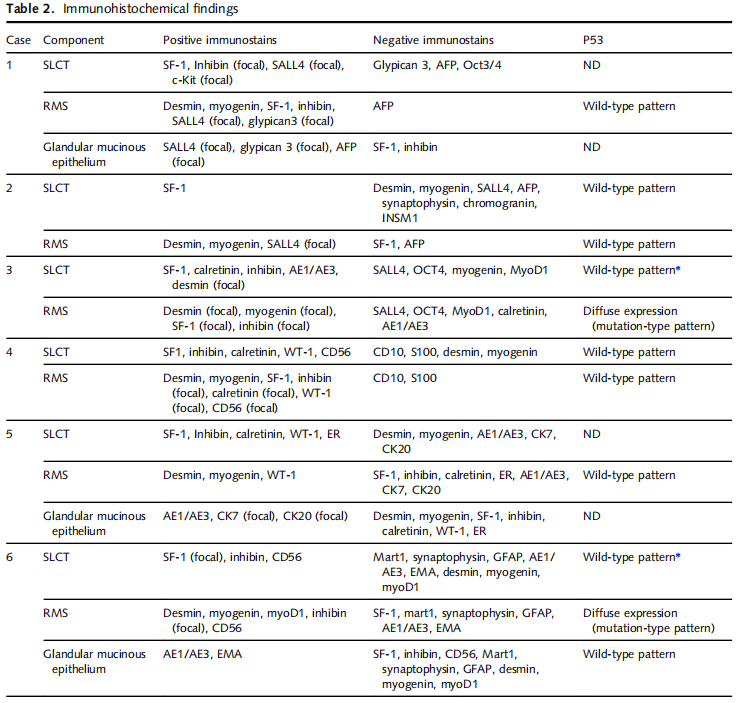
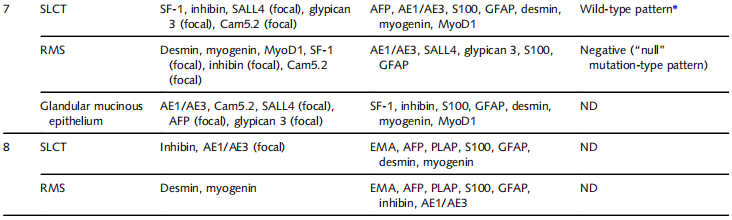
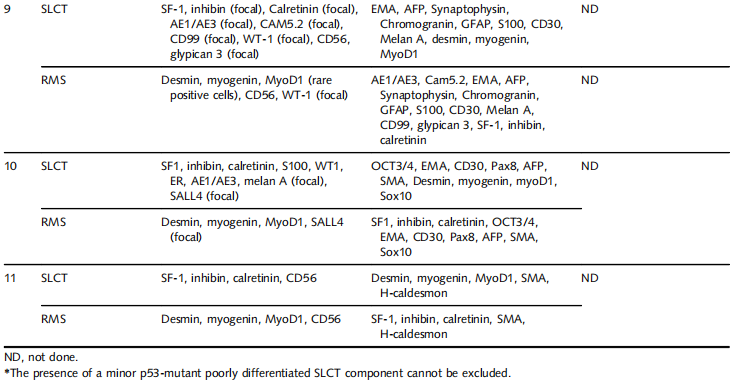
表2
10 例均含第 2 种胚胎型 RMS 成分,由横纹肌母细胞构成,胞质从稀少至丰富嗜酸性不等(病例1和2,图 2A、图 2B),偶见横纹细胞(病例4,图 2C)。束状或结节状梭形细胞呈现分化谱系,从高分化(病例2和4,图 2D、图 2E)至低分化原始细胞伴活跃核分裂象(病例6,图 2F)。多数病例可见细胞稀疏区与密集区交替分布,无 “形成层”,符合无原生上皮的特征。病例 1 部分区域呈纯梭形形态,无明显横纹肌母细胞特征,这类梭形细胞弥漫表达结蛋白及肌细胞生成素。尽管多数病例仅含胚胎型 RMS 成分,部分区域 SLCT 与 RMS 紧密混合(图 2G),SLCT 成分经抑制素及 SF-1 染色凸显(图 2H),RMS 成分经结蛋白及肌细胞生成素染色凸显(图 2I)。病例 6(弥漫核染色)及病例 7(空白型)可见突变型 p53 染色,主要位于肉瘤成分。尽管多数 SLCT 成分呈野生型 p53 表达,因与 RMS 紧密混合,不能排除 p53 突变型低分化 SLCT 成分的存在。所有 10 例形态学及免疫组化特征均支持胚胎型横纹肌肉瘤成分。
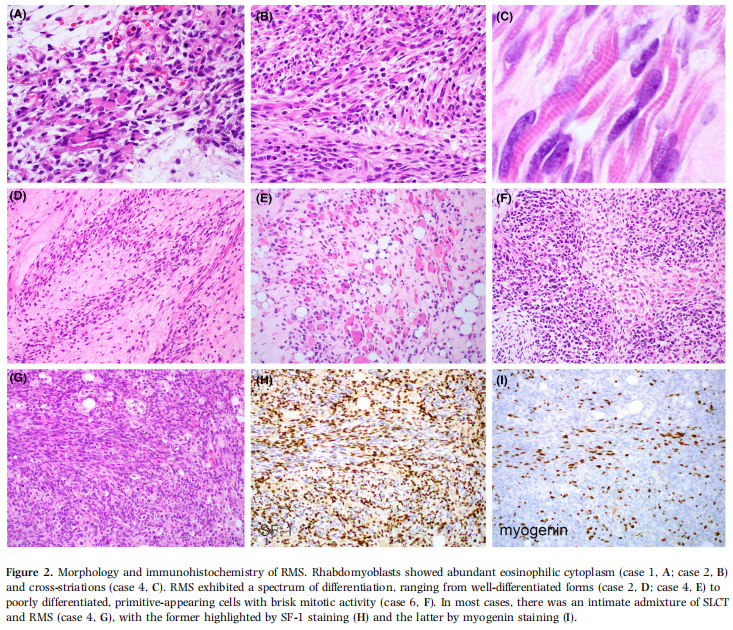
图2
病例 3 与其他肿瘤略有不同,患者 23 岁,因盆腔包块行右侧输卵管卵巢切除术。镜下见中分化 SLCT(图 3A),支持细胞核深染、卵圆形,轻至中度异型性,间质细胞稀疏,多位于肿瘤周边(图 3B)。该成分弥漫阳性表达 SF-1(图 3C)、抑制素及钙视网膜蛋白,p53 呈野生型表达(图 3D)。肿瘤内见多个梭形细胞结节,细胞显著异型,核怪异,核分裂象活跃伴病理性核分裂(图 3E、图 3F)。这类细胞局灶阳性表达结蛋白(图 3G)及肌细胞生成素(图 3H),弥漫核 p53 染色符合突变型(图 3I)。Ki-67 增殖指数在高级别多形性区域显著升高,背景 SLCT 肿瘤区域较低。多形性成分形态及免疫表型符合多形性 RMS。
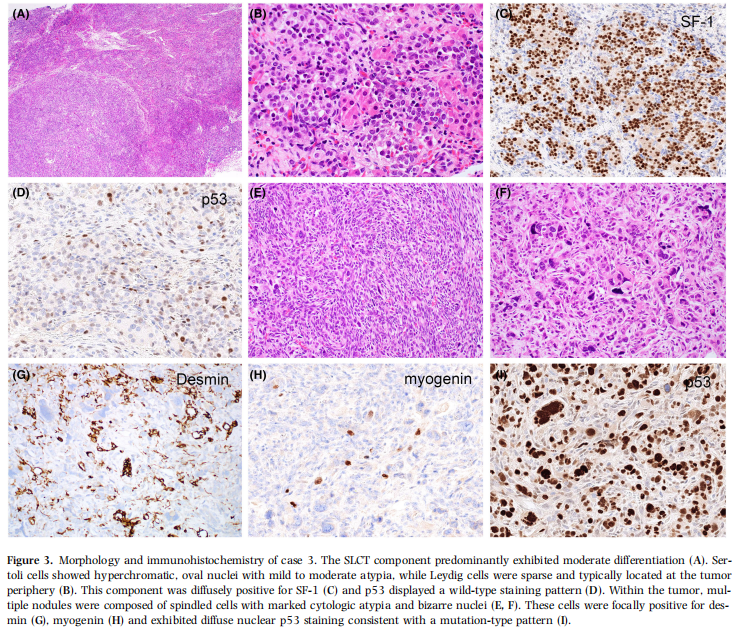
图3
其他异源性成分病理及免疫组化:
除 RMS 成分外,11 例中 8 例(73%)含其他异源性成分。5 例(病例 1、4、5、6、7)见胃肠型黏液上皮(图 4A、图 4B、图 4C),3 例(病例 2、9、10)见细胞学温和的未成熟软骨(图 4D)。其他肉瘤成分包括病例 2 的黏液样梭形细胞肉瘤(图 4E)、病例 5 的上皮样 / 黏液样肉瘤、病例 7 的黏液样多形性肉瘤(图 4F);这类成分均为肿瘤次要成分,视为胚胎型 RMS 的一部分。病例 5 术后 27 个月肿瘤复发并在腹腔内播散,复发 / 转移性肿瘤主要由原始上皮样细胞构成,弥漫表达结蛋白,形态与原发肿瘤中次要的上皮样 / 黏液样肉瘤成分相似。病例 4、5、6 的腺上皮细胞学温和,为典型肠型黏液上皮;病例 1、7 的腺上皮略有不同,可见原始细胞核及胞质透亮(图 5A、图 5B)。这类上皮不表达 SF-1(图 5C),但局灶表达 SALL4(图 5D)、AFP(图 5E)、磷脂酰肌醇蛋白聚糖 3(图 5F),提示肠母细胞分化。
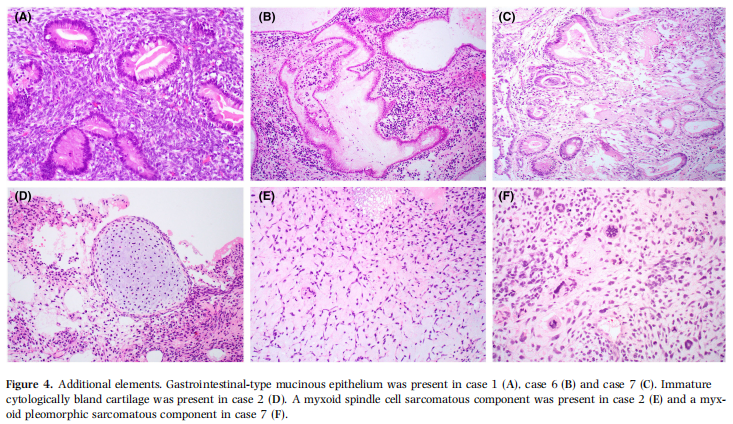
图4
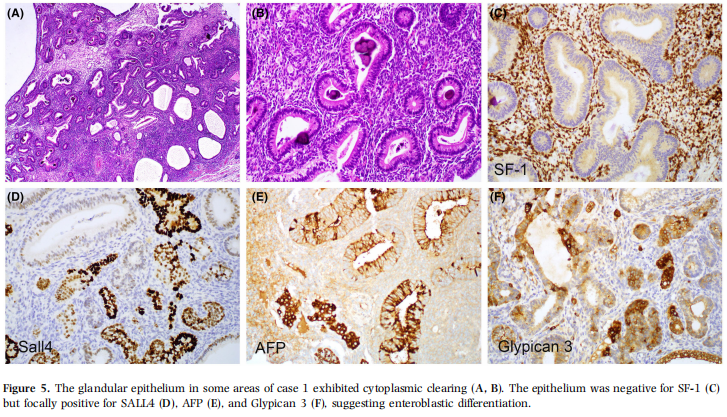
图5
分子变异:
对 7 例患者的 9 份肿瘤组织样本行 NGS 分析,致病性变异及意义未明变异(VUS)总结于表 3 及图 6。所有 7 例患者肿瘤(100%)均携带 DICER1 热点突变,其中 6 例同时存在第 2 个无义或移码功能缺失性 DICER1 突变(图 6A)。病例 3 仅存在 p.D1810Y 热点突变。除 DICER1 突变外,4 例(病例 1、2、4、5)存在 TERT 启动子(c.-124 C>T)突变,3 例(病例 3、6、7)存在致病性体细胞 TP53 突变,与异常 p53 免疫组化结果一致。值得注意的是,TERT 启动子与 TP53 突变在病例中互斥。
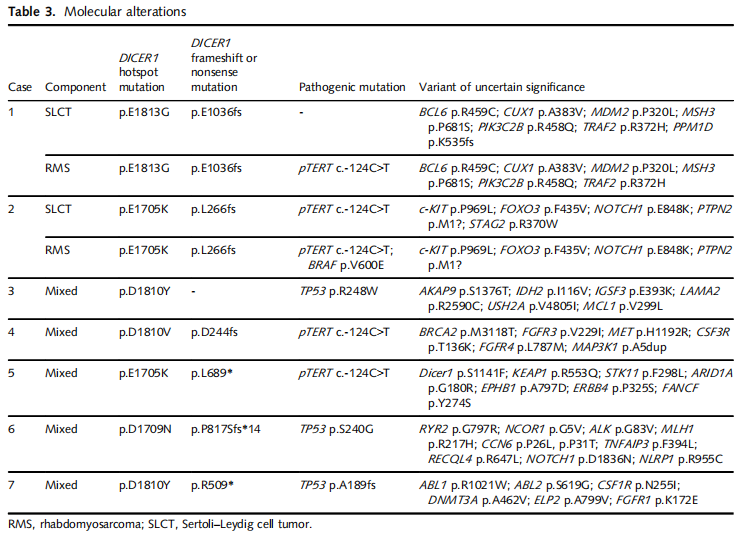
表3
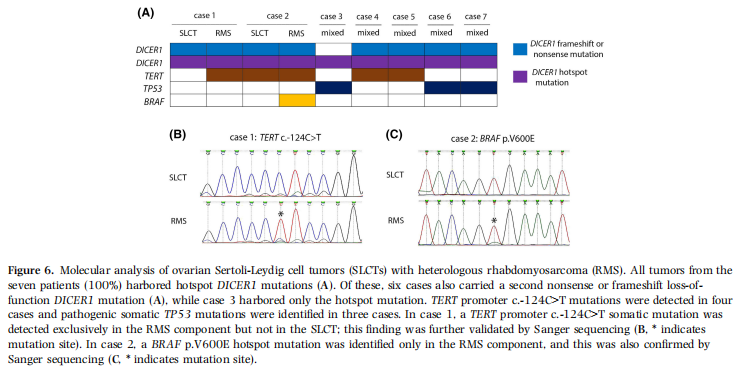
图6
病例 1、2 分别对 SLCT 与横纹肌肉瘤成分行 NGS,比较两种成分分子变异。病例 1 中两种成分均携带 DICER1 p.E1813G 热点突变,支持单克隆起源;TERT 启动子(c.-124 C>T)体细胞突变仅见于 RMS 成分,中分化 SLCT 成分未见,该结果经 Sanger 测序验证(图 6B)。病例 2 中,中分化 SLCT 成分与 RMS 成分共享 DICER1 p.E1705K 热点突变、DICER1 p.L266fs 移码突变及 TERT 启动子(c.-124 C>T)体细胞突变;相反,BRAF p.V600E 热点突变仅见于 RMS 成分,亦经 Sanger 测序验证(图 6C)。尽管仅在少数病例中探究,上述共享及成分特异性分子变异支持 SLCT 与 RMS 成分存在克隆相关性,同时提示额外突变可能参与肉瘤转化。
病例 5 中检测到 8 号染色体三体伴 MYC 扩增。其余病例无明显拷贝数变异,提示可能存在突变等其他机制参与形态学发生,或靶向 NGS panel 检测轻微杂合性缺失、扩增或缺失的敏感性不足。
讨 论
本研究描述了一组伴异源性 RMS 的 SLCT 的临床病理特征及分子变异,这类肿瘤极为罕见,据研究者所知,为本研究单中心报道的最大病例系列,所有病例均为首次报道。所有肿瘤均为单侧,平均大小 22 cm,大于普通 SLCT(平均 12~14 cm),与既往报道的伴异源性软骨和骨骼肌的 SLCT(平均 18.5 cm)相当。根据 2020 年 WHO 女性生殖道肿瘤分类,DICER1 突变型 SLCT 特征为年轻患者、雄激素表现、肿瘤中至低分化、存在网状或异源性成分。本研究所有分子检测病例(100%)均存在 DICER1 突变,整体符合上述特征。但本研究患者平均年龄(34 岁)高于 SLCT 常规报道(平均 25 岁),且呈双峰分布:7 例(64%)≤33 岁(平均 20 岁),4 例(36%)≥52 岁(平均 60 岁)。既往亦有绝经后患者发生伴 RMS 的 SLCT 的报道。40%~60% 的 SLCT 患者存在男性化等雄激素症状,本研究仅 1 例有明确相关表现,多数为会诊病例,雄激素表现细节缺失。
形态学上,本研究所有 SLCT 均为中至低分化;病例 1 约 30% 肿瘤区域可见闭合及空心小管,这类区域偶见于中至低分化 SLCT,不提示高分化成分。SLCT 无论单纯型或伴异源性成分,常呈现多种组织学模式,增加诊断难度。本研究观察到的组织学特征包括弥漫片状、小管、条索状结构、微囊性伴局灶甲状腺样表现、缎带或长条索、网状区域。网状肿瘤患者通常更年轻,中位年龄 15 岁,本研究 2 例局灶网状形态患者分别为 27 岁、71 岁;因网状结构为局灶性,归为伴局灶网状形态的中至低分化 SLCT,而非网状型 SLCT,该区分存在一定主观性。
RMS 是一组显示骨骼肌分化的恶性异质性肿瘤。本研究 10 例存在明确胚胎型 RMS 区域,由分化从成熟至低分化原始的骨骼肌成分构成;另 1 例含梭形细胞区域,细胞显著异型、核怪异,局灶表达结蛋白及肌细胞生成素,符合多形性 RMS 成分。绝经后患者中,SLCT、RMS 及其他上皮和间叶异源性成分并存,易与癌肉瘤鉴别混淆;但 SLCT 特征性形态及支持免疫表型(SF-1、钙视网膜蛋白、抑制素阳性,上皮细胞膜抗原阴性)有助于明确诊断。本研究所有病例中,显示骨骼肌分化的异源性成分呈结节 / 膨胀性肿块,符合 RMS;未发现 SLCT 中散在单个或小群骨骼肌分化细胞(可归类为横纹肌母细胞成分而非 RMS),目前 SLCT 中这类单个或小群细胞应归类为异源性横纹肌母细胞成分还是 RMS 尚不明确。
本研究中,11 例中 3 例(27%)除 RMS 外还含细胞学温和的未成熟软骨。中至低分化 SLCT 中软骨存在(伴或不伴 RMS),以及宫颈胚胎型 RMS 等非性索 - 间质 DICER1 相关肿瘤中胚胎型 RMS 与软骨并存,既往均有报道。这些观察提示肌源性成分(如 RMS)与其他间叶祖细胞(包括参与软骨及广泛间叶分化的细胞)在 DICER1 相关肿瘤中存在复杂相互作用。3 例中存在由梭形和 / 或上皮样细胞构成的黏液样肉瘤灶,进一步支持该理论。既往偶有伴 RMS 的 SLCT 报道神经外胚层成分,本系列未发现。
本研究另一种常见异源性成分为胃肠型黏液上皮,见于 5/11 例(45%)。3 例含形态良性肠黏液上皮,病例 1、7 腺上皮呈现胎儿肠样 / 肠母细胞样形态及免疫表型,特征为上皮细胞核原始、胞质透亮,表达 SALL4、AFP、磷脂酰肌醇蛋白聚糖 3 等肠母细胞标志物。部分病理医师可能将这种肠母细胞分化视为卵黄囊瘤(YST)或 YST 样成分,但该特征既往在卵巢 SLCT 中有报道,研究者认为不应视为 YST 成分。目前无经典 YST 成分与 SLCT 并存的文献报道,不支持这些微小病灶为真正 YST,仍存在争议。笔者之一经验提示,这类伴原始核、胞质透亮、表达生殖细胞标志物的腺体结构是 DICER1 相关肿瘤的另一特征性表现。许多中至低分化 SLCT 伴血清 AFP 升高,本研究数例亦如此。有观点认为异源性肝样细胞或间质细胞可能是血清 AFP 升高来源,也可推测免疫组化 AFP 阳性的肠母细胞成分导致血清水平升高。
笔者之一最初提出宫颈胚胎型 RMS 与卵巢 SLCT 可能存在共同遗传基础的假说,后经分子研究证实。这两种罕见肿瘤目前均被认定为 DICER1 相关肿瘤,存在致病性胚系突变时为 DICER1 综合征表现。但伴 RMS 成分的卵巢 SLCT 分子谱仍未明确。本研究显示所有分子检测病例(7/7,100%)均存在致病性 DICER1 突变,6 例为热点突变 + 无义 / 移码突变,病例 3 仅为热点突变。支持本研究结果,既往 2 例伴 RMS 的 SLCT 亦显示 2 种致病性 DICER1 突变,外周血均未检出,证实为体细胞起源。本研究病例 1、2 成分特异性分析显示 SLCT 与 RMS 成分 DICER1 热点突变完全一致,提示单克隆起源。本研究局限性为缺乏外周血或非肿瘤组织,无法确定这些突变(尤其无义 / 移码突变)为体细胞(散发性)或胚系(综合征性)。尽管本系列缺乏胚系、个人及家族史数据,DICER1 热点突变 + 功能缺失变异并存,结合患者年龄双峰分布,提示部分病例高度可能为胚系 DICER1 综合征。理想情况下,所有伴或不伴异源性 RMS 成分的 SLCT 均应转诊遗传科行胚系 DICER1 突变检测。
尽管存在上述局限,本研究发现了这类独特病理实体的复发性、成分特异性分子变异。本系列 7 例中 4 例(57%)存在 TERT 启动子 c.-124 C>T 热点突变,既往研究 33 例 SLCT 中 2 例(6%)检出相同 TERT 启动子突变,未详细描述临床病理特征。本研究结果提示 c.-124 C>T TERT 启动子突变在伴 RMS 的 SLCT 中富集(57%)。尽管 TERT 启动子突变通常不被视为原发致癌驱动因素,c.-124 C>T 突变与多种恶性肿瘤侵袭性增强、复发率升高、预后更差相关,包括头颈部癌及成人粒层细胞瘤。本系列病例 1 中 TERT 启动子突变仅见于 RMS 成分,中分化 SLCT 成分未见,提示 RMS 可能为肿瘤更高分级成分。相反,病例 2 中 TERT c.-124C>T 突变见于中分化 SLCT 及 RMS 两种成分。仅 2 例对 SLCT 成分单独检测,尚不清楚 TERT 启动子突变是否与 SLCT 分级相关。有趣的是,同一病例中 BRAF p.V600E 突变仅见于 RMS 成分,SLCT 成分未见。BRAF p.V600E 与 TERT 启动子突变并存,既往在甲状腺乳头状癌及黑色素瘤中有报道,已知其提示疾病侵袭性强于单一突变。但病例 2 RMS 成分中两种突变并存是否产生协同效应,导致生物学行为较 SLCT 成分单纯 TERT 启动子突变更具侵袭性,尚不明确。伴 BRAF 突变的胚胎型 RMS 多为 DICER1 野生型、非生殖道来源,病例 2 中该突变存在提示生殖道卵巢 SLCT 伴 RMS 存在独特的分化分子通路,凸显肿瘤多能性及分子多样性。NGS 分析显示 RMS 成分中 BRAF p.V600E 突变等位基因频率低(8.3%),提示为亚克隆变异,进一步支持其为形态演化中的继发事件。但该变异是否驱动独特生物学行为,以及与已知 DICER1 相关与非 DICER1 相关 RMS 谱的差异,仍不明确。
7 例分子分析病例中 3 例(43%)无 TERT 启动子突变,但均存在 TP53 突变,提示这两种突变在这类肿瘤中可能互斥;本研究结果还提示伴 RMS 成分的 SLCT 多数存在 TERT 启动子突变或 TP53 突变。TP53 突变在 SLCT 中罕见,但在胚胎型 RMS 中有报道。因分子分析在 SLCT 与 RMS 混合成分上进行,最初无法确定成分特异性变异。为评估 TP53 突变分布,行 p53 免疫组化,p53 染色模式与 underlying TP53 变异高度相关。发现异常(突变型)p53 表达主要局限于 RMS 成分,尽管不能排除 p53 突变型低分化 SLCT 成分存在。与 TERT 启动子突变结果相似,提示 TP53 突变与 RMS 成分发生相关。研究者推测性索 - 间质祖细胞或多能干细胞获得 DICER1 突变(胚系突变伴体细胞二次打击,或完全体细胞突变),向 SLCT 祖细胞进展;部分细胞维持性索 - 间质谱系形成常规 SLCT,部分细胞累积 TERT 启动子、TP53、BRAF 等额外分子变异,形成 RMS。
本研究病例 3 形态与其他肿瘤略有不同,SLCT 成分主要为中分化,同时见多个结节状高级别肉瘤区域,由梭形细胞及显著异型、常多核、怪异细胞构成,核分裂象活跃伴病理性核分裂。这些区域局灶表达结蛋白及肌细胞生成素,多形性成分形态及免疫表型符合多形性 RMS。分子分析显示该病例存在 DICER1 热点突变及致病性 TP53 突变。值得注意的是,与本研究其他均存在 2 种致病性 DICER1 突变的病例不同,该病例仅存在单一热点突变。免疫组化显示 SLCT 成分呈野生型 p53 染色,Ki-67 增殖指数低;高级别肉瘤区域呈弥漫突变型 p53 表达,Ki-67 指数高。这些结果提示该病例为伴高级别肉瘤转化的 SLCT,符合多形性 RMS。高级别转化现象(非特指多形性 RMS)既往在其他性索 - 间质肿瘤如成人粒层细胞瘤中有报道,TP53 突变及 TERT 启动子突变等分子变异被认为在驱动该转化中起关键作用。尽管组织学特异性突变可能参与形态学演化,鉴于其他存在相似分子事件的性索 - 间质肿瘤可呈现不同组织学形态,可能存在其他机制参与。近期研究显示间变性幼年粒层细胞瘤存在 TP53 突变及 MYC 扩增,部分同时存在 DICER1 突变,有观点认为 MYC 变异在该背景下参与侵袭性行为。本系列中伴 MYC 扩增的病例 5 同样表现出侵袭性行为,是唯一死于疾病的病例,尽管无幼年粒层细胞瘤形态(典型或间变性)。
本研究主要局限性为病例数少、多数病例缺乏随访数据。仅 3 例有随访,均接受化疗,其中 1 例(病例 5)诊断后 27 个月死于转移性疾病,另 2 例随访 12、44 个月无病生存。理想情况下应行原发与复发 / 转移性肿瘤分子变异对比分析,可能揭示肿瘤进展驱动因素,但缺乏转移性肿瘤组织限制了该对比。早期病例系列提示伴异源性未成熟骨骼肌 / RMS 和 / 或软骨的 SLCT 预后与原发性卵巢肉瘤相当,该研究中 10 例患者 8 例术后 5 个月~7 年死于肿瘤相关原因。后续报道同样描述伴 RMS 的 SLCT 患者临床结局不佳。基于这些发现,有观点认为即使诊断时肿瘤局限于卵巢,也应给予辅助化疗,因其潜在侵袭性行为。
综上,本研究报道 11 例伴异源性 RMS 的 SLCT,主要为胚胎型,1 例为多形性。这类肿瘤常含其他异源性成分,所有分子检测病例(7/7,100%)均存在 DICER1 突变,符合 SLCT 的 DICER1 突变分子亚型。除 DICER1 变异外,所有检测病例均存在 TERT 启动子突变或 TP53 突变,二者呈互斥。少数病例成分特异性分析显示这些额外分子变异主要局限于肉瘤成分,提示可能参与这类罕见肿瘤的肉瘤转化。
参考文献:
Zou, Ying S et al. “Ovarian Sertoli-Leydig cell tumors with heterologous rhabdomyosarcoma: Clinicopathologic features and molecular analysis highlighting recurrent genetic alterations.” Histopathology vol. 88,5 (2026): 985-1002. doi:10.1111/his.70058

