靶向急性髓系白血病中的XPO1:从生物学到临床转化
时间:2026-05-09 21:32:33 热度:37.1℃ 作者:网络
深度解析医学证据,lxfs.net为你支撑决策
Exportin-1 (XPO1) 是核质运输的关键调节因子,在急性髓系白血病 (AML) 中经常失调,导致白血病发生、疾病进展和治疗耐药。选择性核输出抑制剂 (SINEs),尤其是塞利尼索和 eltanexor,已显示出有前景的抗白血病潜力,但它们在 AML 中的临床价值、最佳治疗定位和合理使用仍有待充分阐明。
北京医院刘辉教授等近日于《Clinical and Translational Medicine》发表综述,总结了 XPO1 在 AML 中的生物学作用以及 XPO1 抑制剂在临床前和临床环境中的治疗潜力,重点关注 XPO1 的核输出功能及其在 AML 中的致病作用,还总结了 XPO1 抑制剂在 AML 中的作用机制、临床前证据、临床试验结果、不良反应、耐药机制和潜在的反应生物标志物。

本文要点
XPO1 过度激活重新连接核质运输,并在基因定义的急性髓系白血病 (AML) 亚群中维持白血病发生程序。
选择性 XPO1 抑制剂 (塞利尼索, eltanexor) 在 NPM1 突变、DEK::NUP214 阳性和 SF3B1 突变的髓系肿瘤中显示出优先活性。
与去甲基化药物、BCL-2 抑制剂和其他靶向治疗的联合策略增强了缓解的深度和持久性,但受限于毒性。
未来的临床试验应聚焦于分子选择的人群、生物标志物指导的剂量以及转化终点,如可测量残留病 (MRD) 和克隆动力学。
引言
急性髓系白血病 (AML) 具有高复发率和治疗耐药性的特点,在老年患者和有多重合并症的患者中,长期缓解和生存尤其有限。在过去十年中,分子分型、靶向疗法和免疫疗法的进展显著提高了 AML 治疗的精准度和个体化水平。其中BCL-2 抑制剂维奈克拉 (VEN) 与去甲基化药物阿扎胞苷 (AZA) 的低强度联合方案(VEN-AZA,或称 VA 方案)已成为一种新的治疗范式,并被主要国际指南(包括 NCCN、ELN 和 ESMO)推荐为不适合强化化疗的老年患者的一线选择。随机试验和真实世界队列研究表明,VA 方案在这些人群中显著提高了完全缓解 (CR) 和伴血液学恢复不完全的完全缓解 (CRi) 率,并延长了总生存期。然而,大型多中心研究也表明,VA 失败后的挽救选择极其有限,中位总生存期通常少于 1 年,一旦复发通常少于 6 个月。与这些观察一致,作者的单中心真实世界队列显示,初始 CR/CRi 率很高,但中位无事件生存期 (EFS) 约为 9.9 个月,大多数患者在一年内仍出现疾病进展。因此,当代 AML 实践中的一个核心挑战是如何理性地将具有真正新作用机制的疗法整合到现有标准治疗中,以加深缓解、延长缓解持续时间,并为后续的造血干细胞移植创造最佳窗口。
异常核质运输是 AML 发生和发展中的一个重要致病机制。Exportin-1 (XPO1)也称为染色体区域维持蛋白 1 (CRM1),是主要的核输出受体,介导多种肿瘤抑制蛋白和癌症相关信号介质的核输出,包括 p53、FOXO 和 NPM1。XPO1 的过度激活导致肿瘤抑制因子在细胞质中错误定位,致癌转录程序持续激活,并通过转录、表观遗传和翻译网络的多层重新连接,促进白血病细胞增殖、凋亡逃逸和克隆进化。靶向这一节点转运蛋白的选择性核输出抑制剂 (SINE) 可共价抑制 XPO1 活性,并恢复肿瘤抑制通路的核定位和功能。塞利尼索是首个进入临床使用的靶向 XPO1 的 SINE,已获批用于治疗复发或难治性多发性骨髓瘤和弥漫性大 B 细胞淋巴瘤;然而其在 AML 中的最佳治疗定位、获益亚群和合理的联合策略仍在积极研究中。
本综述中首先总结了 XPO1 在 AML 中的生物学功能以及失调的核输出导致白血病发生和疾病进展的机制,特别关注特定的分子亚型。然后回顾了靶向 XPO1 的 SINE 化合物在 AML 中的近期临床前和临床进展,重点关注基因型定义的亚群以及与去甲基化药物、BCL-2 抑制剂和其他通路导向疗法的联合应用。本综述主要基于过去 5 年发表的关于 XPO1 抑制剂在 AML 中的代表性研究,这些研究来自 PubMed 和 Web of Science,以及相关的临床试验注册记录。最后,根据现有的毒性数据和新兴的耐药性见解,讨论了临床实施的关键挑战,并概述了将 XPO1 靶向策略整合到针对生物学和临床上较为复杂的 AML 患者的风险适应性分层和个体化治疗算法中的未来方向。
核输出受体EXPORTIN-1在AML中的生物学作用
核输出蛋白,特别是核胞浆蛋白exportin-1(XPO1),通过调节核质转运,从而控制众多蛋白和 RNA 的亚细胞分布,在维持细胞稳态中发挥核心作用。在 AML 中,XPO1 过表达或失调的 XPO1 依赖性核输出改变了关键肿瘤抑制蛋白和信号介质的定位,破坏了正常的调控网络,并促进了白血病发生和疾病进展。在结构上,XPO1 是一个约 123 kDa 的核转运蛋白-β 家族蛋白,由 21 个串联的 HEAT 重复序列组成,排列成环面状、环形的结构,其 N 端有一个参与 RanGTP 结合的 CRIME 结构域(图 1)。HEAT 重复序列凸起外表面上一个高度保守的疏水沟形成了 NES 结合裂缝,用于容纳货物蛋白上富含亮氨酸的核输出信号 (NES)。RanGTP 的结合稳定了一个“开放”的构象,有利于协同装载含有 NES 的肿瘤抑制蛋白和癌蛋白。SINE 化合物(如塞利尼索和 eltanexor)通过共价修饰 NES 结合裂缝底部的非催化半胱氨酸残基 Cys528 来利用这一结构脆弱性,从而阻断 NES 结合,功能上抑制 XPO1 介导的核输出,并促进关键肿瘤抑制蛋白的核内滞留。
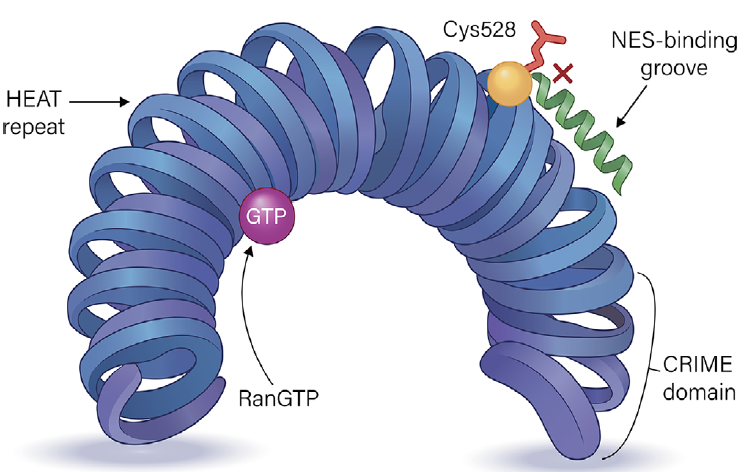
图 1 | 核输出受体 XPO1 的结构组织及 SINE 化合物靶向的 NES 结合裂缝。
XPO1 由 21 个串联的 HEAT 重复序列组成,这些重复序列组装成一个环面状的环形结构,在其凸起的外表面有一个疏水性的 NES 结合裂缝。RanGTP 的结合稳定了开放构象,从而允许装载含有 NES 的货物蛋白。位于 NES 结合裂缝底部的非催化残基 Cys528,是选择性核输出抑制剂(SINEs)的共价靶点,这些抑制剂占据该凹槽,从空间上阻碍 NES 的结合。图中标出了 N 端的 CRIME 结构域。
XPO1 及核质转运机制
XPO1 是主要的核输出受体之一,负责以 Ran GTPase 依赖的方式将数百种蛋白质和多类 RNA 从细胞核输出到细胞质。在细胞核内,Ran 主要以结合 GTP 的形式 (RanGTP) 存在。XPO1 识别带有 NES 的货物蛋白,并与 RanGTP 一起形成三聚体输出复合物。该复合物与核孔复合物 (NPC) 内的核孔蛋白相互作用,穿过 NPC 进入细胞质,在细胞质中,RanGTP 在 Ran GTPase 激活蛋白 (RanGAP) 和 Ran 结合蛋白 (RanBP1/2) 的控制下水解为 RanGDP。水解触发三聚体复合物解离,释放货物,并将 XPO1 回收到细胞核进行下一轮运输。核膜上陡峭的 RanGTP 梯度对于核输出的方向性和保真度至关重要。
除了在间期核质转运中的作用,XPO1还可在有丝分裂过程中定位于动孔和中心体,并促进微管成核、中心体完整性和纺锤体组装,这些功能至少部分独立于其典型的输出活性。在 AML 细胞中,特别是那些具有高增殖和代谢活性的细胞中,XPO1 通常上调,从而增强参与细胞周期控制、DNA 修复和凋亡逃避的货物的核输出(图 2)。
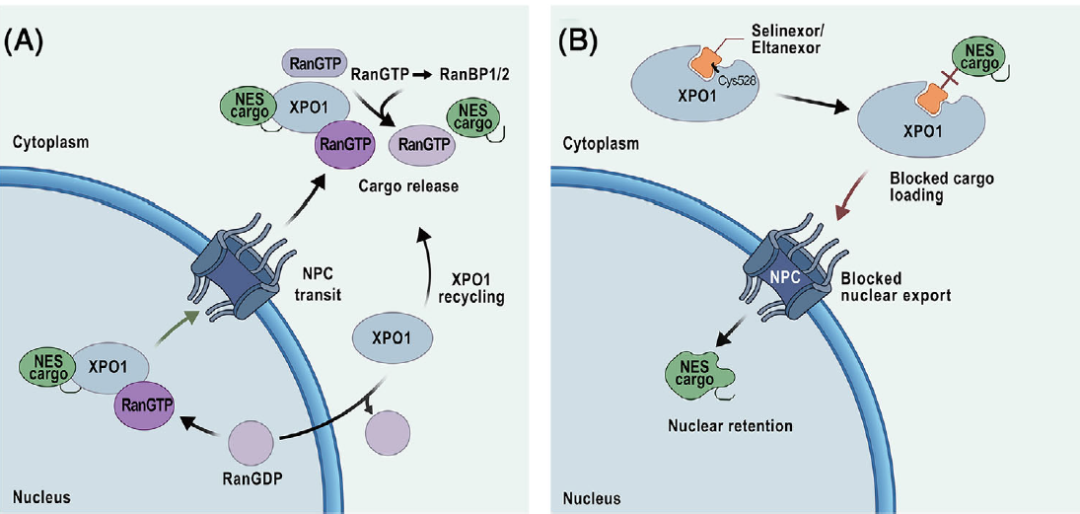
图2
AML中XPO1的失调
在 AML 队列中,XPO1 经常过表达,且高 XPO1 水平与不良预后和治疗耐药性相关。驱动 XPO1 上调的机制尚未完全阐明,但现有数据表明,常见的病变如 MYC 过表达和 TP53 功能障碍可以在转录上促进 XPO1 表达。NES 结合槽内的体细胞突变(例如 E571K)可以改变 XPO1 的构象状态和货物识别谱。尽管此类突变在 B 细胞恶性肿瘤和霍奇金淋巴瘤中更为普遍,但在 AML 中仅罕见地描述了类似的结构改变,其在该疾病中的功能相关性仍不确定。尽管天然存在的 XPO1 突变在 AML 中似乎不常见,但其生物学相关性可能超越简单的突变注释。然而在目前,个体 XPO1 突变在 AML 中的功能和治疗意义仍未充分定义,这些病变最好被视为机制性线索,可能为未来关于 XPO1 依赖性和治疗适应的研究提供信息。
在特定分子亚型中,XPO1 失调与白血病定义性遗传病变之间的相互作用尤为突出。在 NPM1 突变的 AML 中,突变型 NPM1 (NPM1c) 获得了一个新的 NES 基序,并变得依赖于 XPO1 介导的异常胞质输出。此过程将核转录因子 PU.1 及其相关伴侣重新定位到细胞质,破坏与分化相关的转录网络,并维持白血病干细胞样状态。在 FLT3-ITD 阳性的 AML 等高危分子亚型中,XPO1 促进包括 p53 和 FOXO3A 在内的促凋亡因子的核输出,减弱核凋亡信号,并与致癌激酶通路合作以增强细胞存活。
除了 XPO1 本身,输出机制的相关组分,如 Ran、eIF4E、HuR 和 LRPPRC,在包括 AML 亚型在内的几种血液系统恶性肿瘤中也会上调,从而加剧核输出失衡,并导致分子异质性和耐药性。
与AML相关的关键XPO1货物
XPO1 介导多种蛋白质和 RNA 的核输出,这些货物的异常输出在 AML 的发病机制和进展中起着关键作用。 广义上,XPO1 货物可分为两大类。第一类包括肿瘤抑制蛋白,如 p53、RB1、FOXO1/FOXO3A、APC、BRCA1/2、p21 和 p27,这些蛋白在细胞质中的滞留导致其核功能丧失,从而促进不受控制的增殖和凋亡抵抗。第二类包括致癌蛋白和信号调节因子,其异常的核质分布可能支持恶性行为,如 cyclin B1、cyclin D1、SNAIL、TERT、survivin、拓扑异构酶 IIα (TOP2A)、c-ABL 和 YAP1。这些致癌货物的异常胞质积累增强了下游转化信号,并支持白血病细胞的生长和存活。
在包括 AML 在内的血液系统恶性肿瘤中,XPO1 还与 eIF4E-LRPPRC 复合物合作,以增强特定致癌 mRNA 的核输出和翻译。eIF4E 识别含有 eIF4E 敏感性元件 (4ESE) 的转录本,并通过 LRPPRC 和 XPO1 介导其选择性输出。这些转录本包括关键的抗凋亡因子,如 BCL2 和 MCL1,其增强的输出和翻译显著加强了细胞质中的促生存信号。MDM2 是另一个 XPO1 货物,其核输出进一步抑制了 p53 介导的肿瘤抑制活性。此外,CRM1 依赖的 TOP2A 核输出已在几种肿瘤模型中被证明与基于蒽环类药物的化疗耐药性相关。
除蛋白质外,XPO1 还输出多种 RNA 物种和核糖核蛋白 (RNP) 复合物,包括参与核糖体生物发生的核糖体前 RNP、pre-mRNA 剪接所需的 U snRNA,以及由 eIF4E-LRPPRC-XPO1 轴控制的选定癌症相关 mRNA。在 SF3B1 突变骨髓增生异常综合征 (MDS)/AML 中,初步数据表明此类克隆可能对 XPO1 抑制特别敏感,尽管 XPO1 介导的 RNA 输出与剪接体功能障碍之间的直接分子联系仍有待完全阐明。
XPO1 对白血病发生和 AML 病理生物学的贡献
在 AML 中,XPO1 的过度激活通过功能上灭活核肿瘤抑制蛋白并驱动致癌货物在细胞质中积累,促进白血病发生和疾病维持。在 NPM1 突变的 AML 中,XPO1 介导的 NPM1c 及其伴侣 PU.1 的输出改变了核转录复合物的组成和定位。结果,涉及 PU.1、CEBPA 和 RUNX1 的复合物被错误引导,导致分化相关基因的抑制和 HOX/MEIS1 程序的持续激活,从而增强了白血病干细胞样特性。在高风险分子亚型中,包括 FLT3 突变的 AML,XPO1 依赖的促凋亡因子核输出减弱了核死亡信号,并与致癌激酶驱动的增殖通路合作,促进白血病细胞存活和扩增。
除了这些基因特异性效应外,失调的 XPO1 活性还可驱动表观遗传扰动,增强核糖体生物发生,并增加致癌 mRNA 的翻译,从而在多个调控水平上维持 AML 的分子异质性和耐药表型。临床前研究表明,XPO1 抑制可恢复多种肿瘤抑制通路的核定位和功能,诱导白血病细胞凋亡和分化(例如,在 NPM1 突变的 AML 中促进单核细胞分化),并且可能通过依赖 p53 和部分不依赖 p53 的机制发挥作用。AML 研究表明,完整的 TP53 功能与对塞利尼索更高的敏感性相关。
总之,这些发现将 XPO1 确定为 AML 中转录控制、凋亡信号、表观遗传调控和翻译程序的主要调节因子,支持其在白血病起始和维持中的核心作用(图 3)。
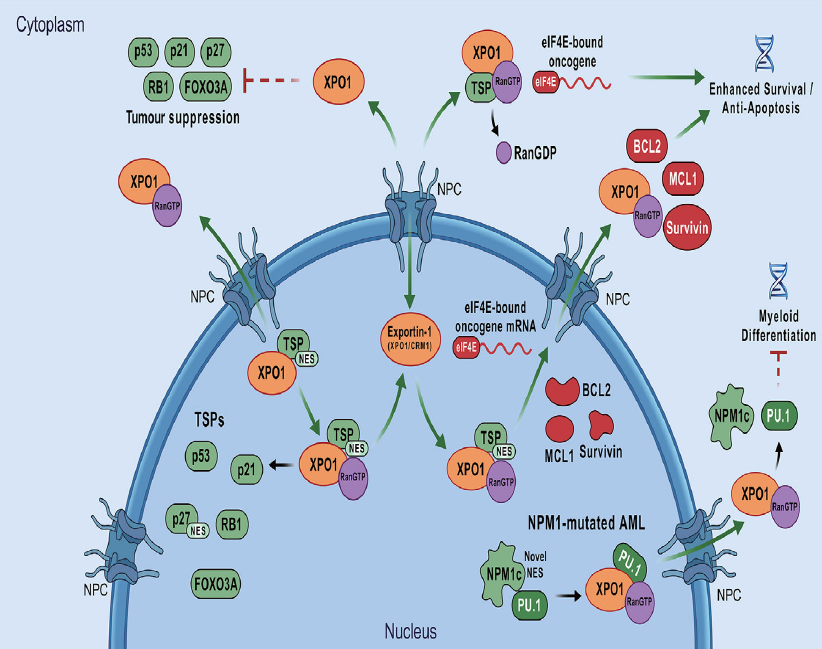
图3
XPO1在AML中靶点特异性作用的遗传学证据
目前,基于 AML 模型中 XPO1 敲除或基因消融的直接证据仍然相对有限,但在几个分子定义的亚型中已出现支持性发现。Charles Cano 等人在 DEK::NUP214 阳性的 AML 模型中显示,XPO1 的缺失显著损害白血病细胞的存活,并诱导细胞周期停滞和凋亡;在患者来源的异种移植模型中,XPO1 抑制也延缓疾病进展并减少白血病负荷。进一步的机制分析表明,XPO1 与 DEK::NUP214 在染色质上共定位,而 XPO1 缺失或药物抑制会破坏这种异常的转录状态,并下调与细胞周期进程和自我更新相关的程序。总之,这些发现表明,至少在特定的分子 AML 亚型中,XPO1 不仅仅是一个药物结合靶点,而是维持白血病状态所需的一个功能性节点。
除了直接的基因消融研究外,支持 XPO1 靶点特异性的另一条重要证据来自遗传学上的靶点验证。Neggers 等人利用基因编辑策略证明,XPO1 中关键药物结合残基 Cys528 的改变赋予了对 塞利尼索 的显著耐药性,并减弱了核输出抑制的典型表型。这些发现提供了遗传学证据,表明 SINE 化合物的核心细胞效应高度依赖于与 XPO1 内特定残基的结合。在 AML 中,这为将塞利尼索和 eltanexor 的抗白血病效应归因于靶向 XPO1 抑制提供了重要的正交支持,并进一步加强了相关临床前研究的机制解释。
总之,这些发现支持了这样一种观点,即 XPO1 不仅仅是一个广泛可成药的核输出因子,而是在特定分子背景下,一个经过遗传和机制验证的白血病维持依赖性靶点。
XPO1抑制剂AML中的临床前研究
SINE 化合物通过与 NES 结合裂缝中的一个关键半胱氨酸残基形成缓慢可逆的共价键来抑制 XPO1,从而阻断 NES 依赖的核输出,并同时干扰与白血病发生相关的多个信号通路。第一代药物如塞利尼索 (KPT-330) 确立了在 AML 中靶向 XPO1 的可行性,而通过结构活性优化开发的第二代抑制剂 eltanexor (KPT-8602) 旨在保持抗白血病效力的同时提高耐受性。最近的临床前研究进一步阐明了 XPO1 抑制在 AML 中的抗白血病效应,包括其下游信号传导后果、在分子定义的 XPO1 依赖性亚群中的活性,以及合理联合策略的潜力。本节讨论的两种主要 SINE 化合物 塞利尼索 和 eltanexor 的化学结构如图 4 所示。
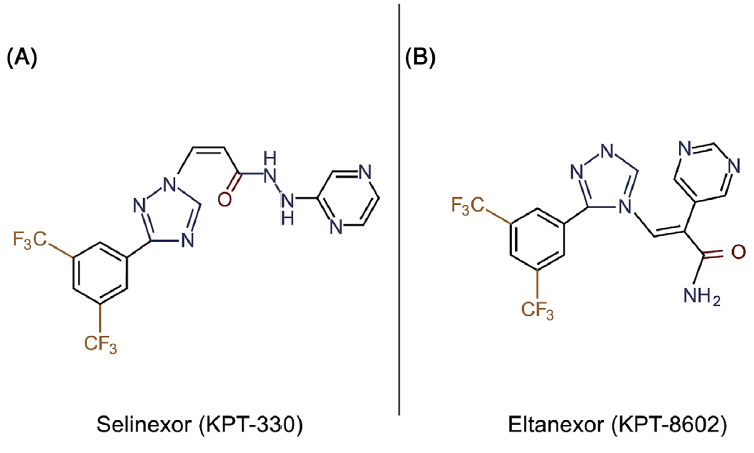
图 4 | 选择性核输出抑制剂(SINEs)塞利尼索和 eltanexor 的化学结构。(A)塞利尼索 (KPT-330)。(B)Eltanexor (KPT-8602)。
塞利尼索 (KPT-330)
塞利尼索是首个进入临床开发并在肿瘤学领域获得监管批准的口服生物可利用的 XPO1 抑制剂。通过共价结合 XPO1 的 Cys528,塞利尼索可阻断转运蛋白与含有 NES 的货物相互作用,并促进包括 p53、FOXO3A 和 RB1 在内的多种肿瘤抑制蛋白的核内滞留,从而恢复其转录和细胞周期调控功能。早期研究表明,塞利尼索可在广泛的 AML 细胞系和原始细胞中诱导细胞周期停滞和凋亡,并在 NSG 异种移植模型中显著减少骨髓白血病负荷,同时相对保留正常的造血干细胞和祖细胞 (HSPC),表明具有有利的治疗指数。
基于这些结果,Emdal 等人对体外塞利尼索处理的原代 AML 样本和细胞系进行了系统的磷酸化蛋白质组学分析。在对塞利尼索敏感的样本中,p53通路组分的磷酸化显著增加,伴随着 p21 等下游细胞周期抑制因子的上调,以及参与增殖和细胞周期进程的蛋白质的去磷酸化。这些数据表明,完整的 p53 功能是产生强效塞利尼索反应的重要先决条件。相比之下,在低反应者或耐药样本中,AKT-FOXO3 通路仍然高度活跃,并部分抵消了 p53 介导的促凋亡信号;AKT 的药物抑制增强了塞利尼索的细胞毒性。这些发现从全局信号层面表明,塞利尼索在 AML 中的抗白血病效应与 p53 通路激活和生存信号抑制密切相关,从而为与 PI3K/AKT 通路抑制剂的联合提供了机制依据。
在 NPM1 突变的 AML 中,XPO1 抑制具有特别令人信服的生物学原理。突变型 NPM1 (NPM1c) 和一些 NPM1 融合蛋白获得了一个新的 NES 基序,该基序以 XPO1 依赖的方式驱动异常的细胞质定位,并维持 HOX/MEIS 驱动的白血病转录程序。Pianigiani 等人在 NPM1 突变的细胞系和患者来源模型中显示,持续的塞利尼索暴露导致 HOXA9、HOXA10 和 MEIS1 的稳定下调,并诱导单核细胞分化标志物如 CD11b 的表达,而间歇性给药仅产生短暂的 HOX 抑制和有限的分化。这些发现表明,在 NPM1 突变的 AML 中,需要持续的 XPO1 抑制才能实现对 HOX/MEIS 程序的持久抑制和终末分化的诱导。
Shimosato 等人将这些观察扩展到罕见的 NPM1 融合亚型,包括 NPM1::MLF1 和 NPM1::CCDC28A。利用小鼠骨髓转导模型和永生细胞系,作者证明这些融合蛋白也作为 XPO1 依赖的 HOX 基因转录激活因子发挥作用。塞利尼索处理减少了 NPM1 融合蛋白与 HOX 启动子的结合,下调了 HOX 表达,并损害了克隆形成生长,支持 XPO1 抑制在 NPM1 融合驱动的 AML 中的潜在作用。
塞利尼索也已在小鼠 AML 模型中与细胞毒性化疗联合进行评估。在一个基因定义的 C57BL/6J AML 模型中,塞利尼索(例如,15 mg/kg 口服,每周三次)联合阿糖胞苷(200 mg/kg 皮下注射)比任意单药更有效地延长中位生存期,并减少外周血和骨髓中的白血病负荷。然而该联合方案对白血病和正常造血均产生抑制作用,表现为 LSK HSPC 和共同淋巴祖细胞显著减少,并在二次移植试验中损害了长期植入能力。这些结果强调,在设计基于 塞利尼索 的联合方案时,需要在增强抗白血病活性和保留正常造血储备之间取得平衡。
Eltanexor (KPT-8602)
Eltanexor (KPT-8602) 是通过对塞利尼索进行结构优化而开发的第二代 SINE,旨在保持抗白血病活性的同时提高耐受性。它保持了对 XPO1 的高亲和力和相似的共价作用机制,但表现出较低的中枢神经系统暴露和更有利的药代动力学及组织分布特征,允许更频繁的给药并可能改善胃肠道耐受性。在高危成人 AML 患者来源异种移植 (PDX) 模型中,Etchin 等人表明,在复杂核型 (AML-CK) 和核型正常 (AML-CN) 模型中,eltanexor 比塞利尼索能更深入地清除骨髓人 CD45+ 白血病细胞,并能更有效地降低白血病起始细胞 (LIC) 的频率,同时对移植了人 CD34+ HSPC 的 NSG 小鼠的正常造血重建产生相对有限的长期影响。62 这些发现支持 eltanexor 作为一种可能在 AML 中更有效、耐受性更好的 XPO1 抑制剂,在临床前模型中对正常造血重建的长期影响相对有限。
最近的研究已经确定了 eltanexor 可能具有特别强效疾病修饰潜力的分子亚型。Charles Cano 等人研究了伴有 t(6;9)(p23;q34)/DEK::NUP214 的 AML,发现 DEK::NUP214 阳性的 FKH-1 细胞高度依赖 XPO1 存活。67 XPO1 的基因消融诱导了深刻的细胞周期停滞和凋亡,而使用 eltanexor 的药物抑制破坏了 XPO1 与 DEK::NUP214 之间的核内和染色质相关相互作用,并在原发性 DEK::NUP214 阳性样本中增加了伴随 G2/M 期积累的凋亡。在 PDX 模型中,eltanexor 治疗导致残留骨髓疾病的深度清除并显著延长了生存期。机制上,染色质免疫沉淀和测序分析表明,DEK::NUP214 和 XPO1 共同占据 HOXB8、HOXB9、PBX3 和其他自我更新相关基因的启动子;eltanexor 破坏了这种染色质相关复合物,下调了 HOX/MEIS 和干性程序,并损害了白血病自我更新。
Kaya 等人在 57 例 DEK::NUP214 阳性 AML 病例(包括 4 例经典 t(6;9))的多组学队列中进一步验证了 XPO1 依赖性。通过使用超过 500 种化合物进行大规模离体药物敏感性分析,他们表明 XPO1 抑制剂 (塞利尼索和eltanexor) 在 t(6;9) AML 中属于最具选择性和最有效的化合物之一。RNA 测序揭示 FOXC1 和 HOXA/HOXB 簇显著上调,而 CUT&RUN 实验证明了 DEK::NUP214 直接结合到 FOXC1 和 HOX 启动子。XPO1 抑制减少了 DEK::NUP214 在这些位点的占据,并下调了相关的转录程序,证实了 t(6;9) AML 中存在一个关键的 DEK::NUP214-XPO1-HOX/MEIS 轴,并为在这种高危亚型中使用 eltanexor 提供了强有力的分子依据。
在 NPM1 突变的 AML 中,Pianigiani 等人系统地评估了 eltanexor 的给药方案。在体外,对 NPM1 突变的细胞系(如 OCI-AML3)连续暴露于 50 mM eltanexor,每周 5 天,可诱导 HOXA9、HOXA10 和 MEIS1 的稳定下调以及 CD11b 的强效上调,而短期或间歇性暴露仅产生短暂的 HOX/MEIS 抑制。在 NPM1 突变的 PDX 模型中,eltanexor(10 mg/kg,每周 5 天,持续 4 周)显著减少了骨髓中人 CD45+ 白血病负荷并延长了生存期,同时伴随着 CD11b 表达增加和持续的 HOX 通路抑制。这些数据表明,持续的 XPO1 抑制对于在 NPM1 突变的 AML 中实现持久的 HOX 簇抑制和诱导终末分化至关重要,并且与基于 塞利尼索 的研究观察结果一致。
在剪接体突变的疾病中,Chaudhry 等人研究了 SF3B1 突变的高危 MDS/AML 模型,包括条件性 SF3b1*K700E 敲入小鼠。Eltanexor 诱导poly(A) + RNA广泛的核保留和异常的选择性剪接,特别是参与细胞凋亡调节和 RNA 加工的转录本。基于 CRISPR 的向前遗传筛选确定 BCL2 和 BCL-XL 为关键的协同节点,体内实验证实 eltanexor 联合维奈克拉优先耗竭 SF3B1 突变的克隆,且具有可接受的全身毒性。这些发现为在 SF3B1 突变的 MDS/AML 中基于 eltanexor 联合 BCL2 抑制的精准医学策略提供了强有力的临床前依据。
联合策略:来自临床前模型的证据
由于 XPO1 抑制同时影响 p53 通路、BCL2 家族成员、MYC 和 HOX/MEIS 驱动的程序以及多个应激反应网络,合理联合已成为 SINE 化合物在 AML 中转化开发的主要焦点。目前的临床前研究主要探讨了与 BCL-2 抑制剂、表观遗传和转录调节剂以及代谢调节剂的联合应用。表 1 总结了 AML 中 XPO1 抑制的关键临床前研究概况,包括疾病模型、分子亚型、联合用药伙伴和机制性见解。
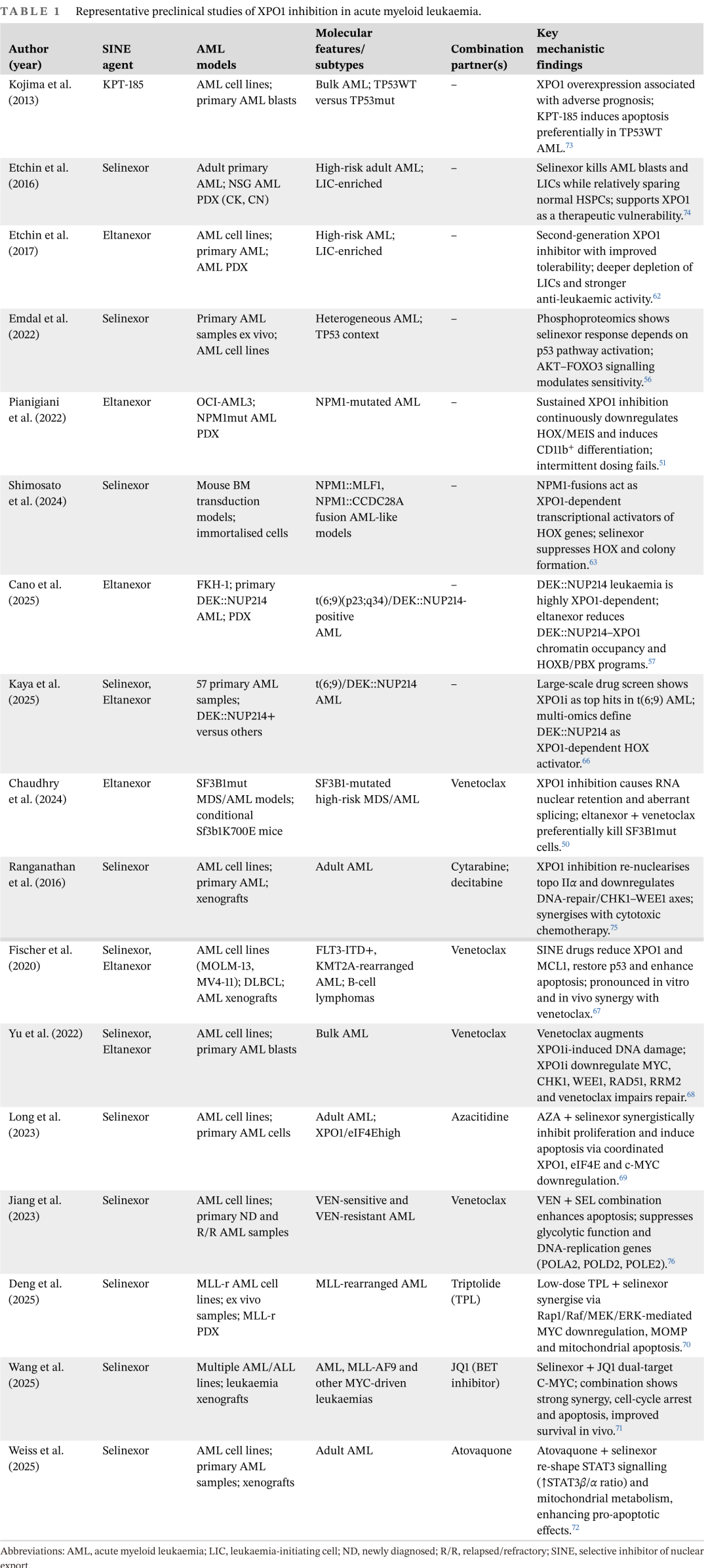
在 BCL2 导向的联合方案中,Fischer 等人系统地评估了维奈克拉与 SINE 化合物 (塞利尼索 和 eltanexor) 在 AML 和弥漫性大 B 细胞淋巴瘤模型中的相互作用。在 AML 细胞系如 MOLM-13 和 MV4-11 中,剂量反应矩阵分析一致显示出协同作用,联合治疗比任一单药产生更大的细胞活力降低和更高的 Annexin V 阳性率。机制研究表明,SINE 治疗下调 XPO1 和 MCL1,恢复 p53 活性,并使线粒体对凋亡敏感,而 维奈克拉 进一步降低了抗凋亡阈值。集落形成试验显示,联合治疗对克隆形成能力的抑制更为显著。在 AML 异种移植模型中,eltanexor(例如,7.5 mg/kg)联合维奈克拉(25 mg/kg)比单药治疗更有效地减少了骨髓和脾脏中人 CD45+ 白血病浸润,并延长了生存期。
Yu等人从DNA损伤的角度探讨了这种联合策略,发现塞利尼索单药治疗会增加γH2AX水平,这与DNA双链断裂的积累相一致,而加入维奈克拉则能进一步提高γH2AX水平,同时下调RAD51和CHK1等DNA修复蛋白。RRM2和WEE1的表达也出现下调,从而削弱了DNA损伤反应,降低了细胞对基因毒性应激的耐受性。在AML模型中,与任一单药相比,联合治疗使细胞活力额外降低了40%-50%。在SF3B1突变的MDS/AML模型中,前述Chaudhry等人的研究表明,与单独使用eltanexor相比,eltanexor联合维奈克拉能更有效地抑制SF3B1突变的克隆,这进一步支持了以下观点:XPO1抑制联合BCL2抑制可能代表一种基于基因型的联合策略,而非纯粹的经验性联合。
与表观遗传和转录调控因子的联合也显示出潜力。Long等人报道,阿扎胞苷联合塞利尼索在AML细胞模型中能协同抑制增殖并诱导凋亡。其机制在于,阿扎胞苷减弱了c-MYC驱动的致癌转录程序,而塞利尼索则破坏了XPO1/eIF4E依赖的MYC及其靶标转录本的核输出和翻译,从而对MYC轴产生多层抑制。在KMT2A(MLL)重排的AML中,Deng等人发现,低剂量的雷公藤甲素联合塞利尼索能显著增强线粒体凋亡。在细胞系和PDX模型中,这种联合方案抑制了Rap1/Raf/MEK/ERK信号级联反应,下调了MYC,降低了线粒体膜电位,增加了活性氧(ROS)的积累,并升高了γH2AX水平,从而放大了DNA损伤并触发凋亡。Wang等人进一步证明,塞利尼索联合BET抑制剂JQ1在多种AML细胞系和白血病异种移植模型中发挥协同抗白血病效应,在相对较低的剂量下实现超过85%的生长抑制,联合指数<1,并更显著地下调C-MYC,增加cleaved PARP和细胞周期阻滞。71 在MLL-AF9驱动的CDX/PDX模型中,塞利尼索联合JQ1显著减少了骨髓、脾脏和肝脏中的白血病浸润,并延长了生存期。总之,这些研究强调,XPO1抑制剂可以通过集中靶向MYC和HOX/MEIS致癌轴,与去甲基化药物、BET抑制剂及其他转录调节因子协同作用。
在代谢及其他非经典联合方案领域,Weiss等人报道,抗疟药物阿托伐醌在AML细胞系、原代样本和异种移植模型中与塞利尼索具有协同作用。在体外,该联合方案比任意单药更能有效抑制细胞活力并诱导凋亡。机制分析表明,阿托伐醌通过抑制电子传递链来干扰线粒体氧化磷酸化,而塞利尼索则阻断XPO1介导的核输出;两者共同作用,提高了STAT3β/α的比率,并上调了下游靶点如SELL(CD62L)的表达,从而重塑了STAT3信号传导,增强了抗白血病效应。在异种移植模型中,阿托伐醌联合塞利尼索减少了骨髓白血病浸润并延长了生存期,而较高的CD62L表达与更好的预后相关。这些发现表明,将XPO1抑制与靶向能量代谢的药物相结合,可能为复发或难治性AML提供更多的联合治疗机会。
总体而言,目前的临床前证据表明,特定的分子亚型(包括 NPM1 突变型、t(6;9)/DEK::NUP214 阳性和 SF3B1 突变型疾病)表现出对 XPO1 的高度依赖性,并且可能对 XPO1 抑制特别敏感。在联合策略方面,将 XPO1 抑制剂与 BCL-2 抑制剂、去甲基化药物、BET 抑制剂或代谢调节剂配对的方案,已在不同的 AML 模型中显示出协同抗白血病活性,并得到合理的机制原理支持。未来转化工作的关键问题包括:如何在控制血液学和全身毒性的同时,维持足够强效和持续的 XPO1 抑制;以及如何根据分子亚型和信号通路依赖性选择最佳的联合伙伴。
XPO1 抑制在 AML 中的临床研究
尽管 XPO1 抑制剂已在多种 AML 模型中显示出明确的抗白血病活性,但在 AML 中的临床转化仍处于早期阶段。现有数据主要来自塞利尼索在复发/难治性 (R/R) 疾病中的 I/II 期试验,以及第二代抑制剂 eltanexor 在原始细胞过多的高危 MDS 中的研究。总体而言,塞利尼索单药治疗在 R/R AML 中产生的缓解率不高;尽管与强化化疗或低强度方案的联合可以提高初始缓解率,但毒性与生存获益之间的平衡是变化的,XPO1 抑制的最佳临床定位仍有待确定。表 2 提供了 XPO1 抑制在 AML 和相关高危髓系肿瘤中的关键临床研究概况。
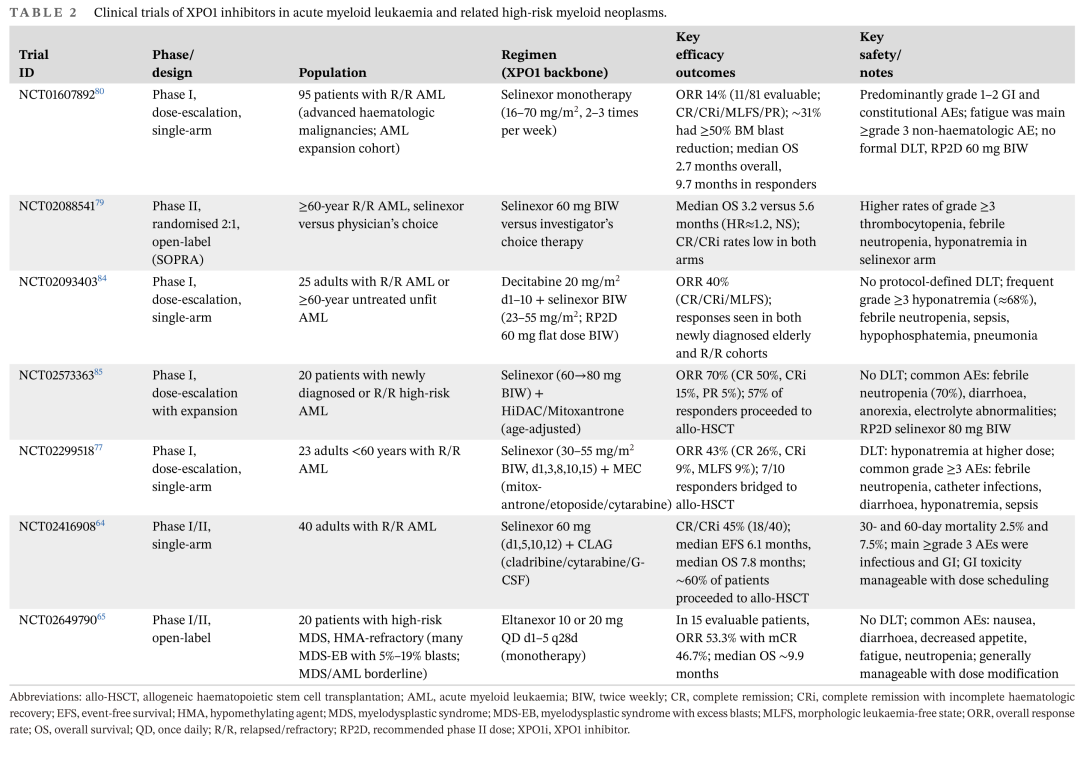
塞利尼索单药治疗AML
在随机、开放标签的 II 期 SOPRA 试验 (NCT02088541) 中,Sweet 等人将年龄 ≥ 60 岁的 R/R AML 患者以 2:1 的比例随机分配接受塞利尼索60 mg 每周两次或研究者选择的治疗(n = 118 vs. 57)。塞利尼索组的中位总生存期 (OS) 为 3.2 个月,而医生选择组为 5.6 个月(HR ≈ 1.18, p=0.422),两组的 CR 或 CRi 率均较低。塞利尼索组≥3 级血小板减少、发热性中性粒细胞减少和低钠血症更常见。作者得出结论,在未经选择的老年 R/R AML 人群中,塞利尼索单药治疗具有有限的抗白血病活性和显著的毒性负担,更适合作为联合策略的骨架,而非作为单一疗法。
在早期的 I 期剂量递增研究 (NCT01607892) 中,Garzon 等人用塞利尼索治疗了 95 例 R/R AML 患者,剂量范围广泛。80 在 81 例可评估患者中,总缓解率(CR/CRi/骨髓无白血病状态 (MLFS)/部分缓解)为 14% (11/81),约 31% 的患者骨髓原始细胞减少 ≥ 50%。整个队列的中位 OS 为 2.7 个月,而有反应者的中位 OS 接近 9-10 个月,表明部分高危患者可以获得相对持久的疾病控制。综上所述,目前的单药治疗数据表明,塞利尼索在重度经治的 R/R AML 中疗效一般,其最有前景的作用可能是在合理设计的联合方案中,而非作为单药标准治疗。
塞利尼索联合强化化疗
在 R/R AML 的挽救治疗中,塞利尼索已在多项早期试验中与强化化疗联合。II 期 SAIL 研究 (NCT02249091) 在 R/R AML 患者 (n = 42) 中评估了塞利尼索联合标准的“7+3”方案(阿糖胞苷联合伊达比星)。初始塞利尼索剂量为 40 mg/m² 每周两次,持续 4 周;然而该方案与长期中性粒细胞减少相关,分别有 85% 和 56% 的患者出现发热性中性粒细胞减少和 3-4 级腹泻。随后修订方案为固定剂量的塞利尼索 60 mg 每周两次,持续 3 周,并高了耐受性。在整个队列中,客观缓解(ORR;CR/CRi/MLFS) 为 50%,约三分之一的患者接受了异基因造血干细胞移植 (allo-HSCT)。中位 EFS、无复发生存期 (RFS) 和 OS 分别为 4.9、17.7 和 8.2 个月。这些数据表明,塞利尼索联合“7+3”在部分R/R AML 患者中具有有意义的挽救治疗潜力,但需要仔细优化剂量和方案以平衡疗效与骨髓抑制和胃肠道毒性。
Bhatnagar 等人进行了一项 I 期剂量递增研究 (NCT02299518),在 < 60 岁的 R/R AML 成人患者 (n = 23) 中评估了 塞利尼索联合米托蒽醌、依托泊苷和阿糖胞苷 (MEC)。塞利尼索 的最大耐受剂量为 30 mg/m²,剂量限制性毒性主要包括 3-4 级低钠血症。ORR 为 43%,其中 CR 率为 26%,表明该联合方案具有临床相关的挽救活性,但需要密切监测和支持治疗电解质紊乱和感染相关毒性。
在新诊断的高危或不良风险 AML 中,Sweet 等人在一项 I 期研究 (NCT02403310) 中评估了塞利尼索联合柔红霉素和阿糖胞苷诱导化疗。在推荐的 II 期剂量 80 mg 塞利尼索每周两次下,ORR 约为 70%,CR 率约为 50%,显示出显著的白血病活性。但≥3 级胃肠道不良事件和电解质异常很常见,强调了在一线强化方案中整合塞利尼索时需要优化剂量和进行主动的支持性管理。
重要的是,Janssen 等人进行的一项多中心、开放标签、随机 II 期试验(荷兰试验注册号 NL5748 [NTR5902])在年龄 ≥ 65 岁、适合强化化疗的患者中评估了在标准“3+7”诱导方案中加入塞利尼索的效果(n = 102;每组 51 例)。在这项研究中,对照组达到的 CR/CRi 率为 80%,显著高于塞利尼索组观察到的 59%。对照组的 18 个月 EFS 和 OS 分别为 45% 和 58%,而塞利尼索组分别为 26% 和 33%。塞利尼索组的感染、脓毒症和治疗相关死亡率也更高,且可测量残留病 (MRD) 阴性率没有改善。这些发现强调,在接受一线强化治疗的未经选择的老年患者中,简单加入塞利尼索不仅可能无法改善预后,反而可能恶化耐受性,从而反对将其作为一种普遍的一线强化策略。
塞利尼索联合低强度治疗
在年龄较大或不适合强化化疗的患者中,已探索了塞利尼索与低强度方案的联合。在一项 I 期剂量递增研究 (NCT02093403) 中,Bhatnagar 等人将塞利尼索与低剂量地西他滨 (20 mg/m²) 联合用于 R/R AML 成人患者或年龄 ≥ 60 岁不适合强化治疗的新诊断患者 (n=25)。未观察到方案定义的剂量限制性毒性,推荐的 II 期剂量确定为塞利尼索 60 mg 每周两次。ORR 为 40% (CR/CRi/MLFS),表明该联合方案可以在部分高危患者中诱导缓解。然而≥3 级低钠血症很常见(约 68%),同时发热性中性粒细胞减少、脓毒症、低磷血症和肺炎的发生率也很高。缩短塞利尼索给药持续时间并转换为固定剂量方案在一定程度上改善了耐受性。总体而言,塞利尼索联合低强度去甲基化治疗在部分难治患者中是可行的,但其使用受到电解质紊乱和感染相关毒性的限制。
除了传统的临床终点,将克隆动力学和 MRD 评估相结合可能有助于完善 XPO1 抑制剂联合方案的患者选择。在 SAIL 试验的一项相关子分析中,Klement 等人检查了 15 例接受塞利尼索联合化疗治疗的 R/R AML 患者的克隆演化情况。携带 FLT3、SF3B1 和 TP53 突变的克隆的变异等位基因频率在治疗后显著下降,而涉及与年龄相关的克隆性造血基因(如 DNMT3A、ASXL1 和 TET2)的克隆则保持稳定或略有增加。在现有的随访中,这些模式之间的中位生存差异尚未明确显现。尽管如此,这些观察结果表明,将克隆和 MRD 动力学纳入未来的试验可以提供额外的见解,了解哪些患者能从含 XPO1 抑制剂的方案中获得持久益处,并可能有助于建立临床相关的药效动力学和早期耐药生物标志物,用于治疗监测。
第二代抑制剂 Eltanexor 的临床探索
Eltanexor 是第二代 XPO1 抑制剂,旨在实现比塞利尼索更低的中枢神经系统暴露和更好的胃肠道耐受性,从而实现更频繁或更长时间的给药。尽管 AML 中尚无大型前瞻性试验,但在原始细胞过多的高危 MDS 中的研究提供了重要线索。在一项 I/II 期开放标签试验 (NCT02649790) 中,Lee 等人评估了口服 eltanexor 单药治疗对去甲基化药物原发性难治或复发后高危 MDS 患者的疗效;许多患者骨髓原始细胞为 5%-19%,接近 WHO 2022 分类中的 MDS/AML 边界。在 15 例接受 eltanexor 10 或 20 mg 每日一次、28 天周期第 1-5 天治疗的可评估患者中,ORR 为 53.3%,骨髓完全缓解 (mCR) 率为 46.7%,中位 OS 约为 9.9 个月。未观察到剂量限制性毒性。最常见的治疗相关不良事件为恶心、腹泻、食欲减退、疲劳和中性粒细胞减少,其中大多数可通过剂量调整和支持治疗来控制。
结合在NPM1突变型、t(6;9)/DEK::NUP214阳性以及SF3B1突变的MDS/AML中积累的大量临床前数据,这些发现表明,eltanexor更有可能通过“分子筛选联合持续抑制”的策略在AML中找到其定位,而非通过在未经选择的人群中复制赛利尼索的给药方案来实现。虽然在典型AML队列中的前瞻性数据仍然很少,但在原始细胞过多的高危MDS中观察到的安全性特征和初步疗效,为设计跨越MDS-AML连续谱并聚焦于遗传学和生物学定义亚群的试验提供了理论依据。
XPO1抑制剂的风险与挑战
尽管 XPO1 抑制剂已在临床前模型和早期临床试验中显示出抗白血病活性,但缓解的持久性和安全性仍然是其广泛使用的主要限制。在一项针对接受塞利尼索联合“7+3”治疗的不良风险新诊断 AML 患者的队列中,Sweet 等人报告 CR/CRi 率约为 53%,但中位 OS 仅为约 10.3 个月,表明即使在强化化疗背景下,长期预后仍然有限。在一项将塞利尼索与高剂量阿糖胞苷和米托蒽醌联合的 I 期研究中,总缓解率接近 70%,但仍有部分患者出现快速进展或早期复发。机制研究表明,白血病细胞可以通过多种适应性途径减弱 XPO1 抑制的作用,包括上调 XPO1 本身、重塑更广泛的核质运输机制以及激活应激反应通路,如热休克蛋白、脂质代谢和氧化磷酸化。在一些模型中,TP53 功能状态和 c-MYC/E2F 轴的异常激活也与敏感性降低和获得性耐药相关。总之,这些发现表明,在目前的发展阶段,XPO1 抑制剂更有可能作为机制指导下的组合或序贯方案中的有效组分,以加深和延长对现有疗法的反应,而非作为旨在提供持久长期疾病控制的单一疗法。
从安全性的角度来看,XPO1 抑制剂的毒性特征与其在正常造血和非造血组织中对核输出的广泛抑制密切相关。在将塞利尼索与强化化疗联合的 I 期试验中,最常见的 3-4 级非血液学不良事件包括发热性中性粒细胞减少(高达约 67%)、腹泻(约 29%)、低钠血症(约 29%)和脓毒症(约 14%)。在包含高剂量阿糖胞苷和米托蒽醌的方案中,发热性中性粒细胞减少的发生率约为 70%,同时经常报告腹泻(约 40%)、厌食(约 30%)、电解质紊乱(约 30%)以及疲劳和恶心/呕吐(约 25%)。一项对 FAERS 数据库的大型真实世界药物警戒分析确定了 4392 份将塞利尼索列为疑似药物的不良事件报告。胃肠道毒性约占报告的 23.1%,血小板减少和贫血等血液学异常约占 6.9%,疲劳、体重减轻和头晕等非特异性症状约占 12.9%。值得注意的是,55.3% 的事件与住院相关,28.9% 与死亡相关,突显了发生严重后果的非平凡风险。临床前工作进一步表明,塞利尼索相关血小板减少与早期巨核细胞祖细胞中血小板生成素信号的抑制有关,符合“靶点相关”的机制性毒性。对于通常基线时骨髓储备受损且营养状况不佳的 AML 患者,这些毒性会加剧,常常需要减少剂量、延迟或中断治疗,以及加强支持治疗;这反过来又缩小了实际的治疗窗口,并限制了 XPO1 抑制剂与其他骨髓抑制药物联合使用的强度。
针对这些局限性,第二代 XPO1 抑制剂(如 eltanexor)被设计为具有改变的药代动力学和组织分布特性,以减少中枢神经系统和全身暴露,从而提高耐受性。在一项针对高危、HMA 难治性 MDS 的 I/II 期试验中,许多患者的骨髓原始细胞为 5%-19%,接近 MDS/AML 边界,未观察到正式的剂量限制性毒性,3-4 级不良事件的总体发生率也低于 AML 队列中报道的 塞利尼索。然而,约 35% 的患者需要延迟治疗,约 40% 的患者因毒性需要减少剂量,表明尽管不良事件负担有所减轻,但仍然具有临床意义。因此,AML 领域面临的一个关键挑战是如何在控制血液学和全身毒性的同时,维持药效学上有效且足够持续的 XPO1 抑制。实现这一目标需要优化给药方案、主动毒性管理和开发可预测疗效和不耐受的生物标志物,从而能够合理选择患者并在特定临床背景下个性化应用 XPO1 抑制剂。
转化视角与讨论
AML 的特点是复发率和治疗耐药性高,老年患者和有多重合并症患者的长期缓解和生存尤其有限。基于维奈克拉的低强度方案已成为老年和不适合强化患者的主要治疗选择,并且在随机试验和真实世界队列中,已明确改善了初始缓解率和总生存期。尽管如此,中位 EFS 或 RFS 通常仍在 8-10 个月的范围内,并且复发常伴随多克隆进化和获得性耐药,留下的有效挽救选择极少。因此,将具有真正新作用机制的药物引入这个重度经治的空间已成为当代 AML 实践中的一个核心挑战。
在此背景下,以塞利尼索为代表的 XPO1 抑制提供了一种机制上独特的策略,旨在核质运输水平重新编程白血病信号。塞利尼索目前已获批用于复发/难治性多发性骨髓瘤和弥漫性大 B 细胞淋巴瘤,而其在 AML 中的作用仍在定义中。通过共价靶向 XPO1 中的 Cys528 并阻断肿瘤抑制蛋白和致癌转录调节因子的异常核输出,XPO1 抑制剂在 NPM1 突变或融合 AML、DEK::NUP214 阳性疾病、剪接体突变亚群和某些治疗耐药克隆中具有强有力的生物学原理。临床前研究一致表明,XPO1 抑制可减少白血病负荷,靶向 LIC,并与去甲基化药物、BCL-2 抑制剂和 BET 抑制剂在多种 AML 模型中协同作用,为在高危和复发/难治人群中的临床探索提供了坚实的机制基础。然而AML 中的临床数据仍然有限,主要来自小型、早期或单臂研究,而在明确界定的亚组中的对照证据仍然很少。
XPO1 抑制剂在 AML 中更广泛实施的主要障碍之一是其毒性特征所决定的狭窄治疗窗口。塞利尼索已在 I/II 期试验中显示出抗白血病活性,但这种活性伴随着显著的血液学和非血液学毒性,包括血小板减少、中性粒细胞减少、感染风险增加以及胃肠道不良事件,如恶心、厌食、体重减轻和低钠血症。在骨髓储备已经受损且营养状况不佳的患者群体中,这些毒性通常会被放大,常常需要减少剂量、延迟或中断治疗,以及加强支持治疗。这一现实压缩了持续治疗的实际窗口,并限制了 XPO1 抑制剂与其他骨髓抑制疗法联合使用的强度。这也可能有助于解释为什么塞利尼索未能在 AML 中成功进入更前线的治疗:在一项随机试验中,在适合强化治疗的老年患者的一线“3+7”方案中加入塞利尼索未能改善预后,并且在未经选择的人群中甚至显示出劣于标准治疗的迹象。旨在通过改变药代动力学和组织分布以减少中枢神经系统和全身暴露的第二代药物如 eltanexor,在原始细胞过多的高危 MDS 中显示出更有利的耐受性,但其在典型 AML 队列中的有效性和安全性仍需严谨、系统的评估。
这些考虑表明,XPO1 抑制的临床价值不太可能在于为 AML 提供另一种广泛适用的“一刀切”靶向疗法。恰恰相反,XPO1 抑制剂可能最好在生物学定义的环境和特定的治疗窗口中,作为精心选择的联合或序贯策略的组成部分来使用。未来的工作可以集中在:(1) 具有推定高 XPO1 依赖性的分子亚型,例如 NPM1 突变或 NPM1 融合 AML、t(6;9)/DEK::NUP214 阳性疾病和剪接体突变亚群;(2) 维奈克拉-阿扎胞苷或相关方案治疗失败但仍适合强化挽救治疗或异基因移植的患者;以及 (3) 具有持续性 MRD 或高危克隆进化信号的个体,在这些个体中,当整合到风险适应性路径中时,XPO1 抑制可用于加深反应或延缓进展。在这些情况下,XPO1 抑制剂将作为现有骨干疗法的靶向“附加”药物,而非作为独立的骨干疗法。
在这个转化框架内,XPO1 突变的治疗意义也值得更明确地考虑。与 NPM1 突变或 DEK::NUP214 重排不同,天然存在的 XPO1 突变目前并未定义 AML 中的主要分子亚型,并且似乎相对不常见。尽管如此,这些突变仍然可能具有生物学信息价值,因为 NES 结合区或相关结构元件的改变可能影响货物识别、输出选择性,并可能影响白血病对 XPO1 介导的核输出的依赖程度。然而从治疗角度来看,目前的证据仍不足以支持 XPO1 突变状态作为选择基于塞利尼索或 eltanexor 治疗 AML 的生物标志物。目前,这些病变最好被视为机制性线索,可能有助于完善未来对 XPO1 依赖性、亚型特异性脆弱性和治疗适应的研究,而不是作为常规实践中的临床可操作生物标志物。重要的是,天然存在的 XPO1 突变也应与药物结合残基 Cys528 的实验性改变区分开来,后者主要为 SINE 化合物提供靶点验证和耐药相关证据,而非反映 AML 的复发基因组特征。
更广泛地说,对于 XPO1 抑制剂在 AML 中的临床开发,生物标志物应被概念化为至少三个互补的类别:预测性生物标志物、药效动力学生物标志物和耐药性生物标志物。预测性生物标志物需要在治疗开始前识别出最有可能受益的患者亚群,可能包括与高 XPO1 依赖性相关的分子背景,例如 NPM1 突变或 NPM1 融合 AML、t(6;9)/DEK::NUP214 阳性疾病、SF3B1 突变的髓系肿瘤,以及在某些情况下的P53 完整的信号状态。药效动力学生物标志物需要在治疗期间确认靶点参与和生物学反应,可能包括 XPO1 货物的核内重定位、HOX/MEIS 驱动的转录程序的抑制、分化标志物如 CD11b 的诱导、p53/p21 相关信号的激活以及 MRD 或突变负荷的动态变化。耐药性生物标志物则可能有助于解释原发性难治性或获得性治疗失败,可能包括 TP53 功能障碍、持续的 AKT-FOXO3 或 MYC/E2F 信号、核质运输和应激反应通路的适应性重塑,以及在治疗压力下的克隆持续存在或进化。将这三个生物标志物层面前瞻性地整合到未来的试验中,可以改善患者选择、治疗中监测和复发监测,从而促进 XPO1 靶向策略在 AML 中的更精准临床定位。
同时,联合方案的开发应从经验性堆叠转向基于机制的整合。XPO1 抑制与 BCL2 依赖性、MYC/E2F 驱动的转录、DNA 损伤反应通路和代谢重编程之间新兴的相互作用,为设计在药效学上有效且可耐受的聚焦性联合方案提供了理论基础。优先考虑 MRD 动力学、克隆进化和持久生存终点,而非仅仅短期缓解率的试验,对于识别真正有益的策略至关重要。同时,生物标志物的开发也至关重要。未来的生物标志物框架不应仅仅依赖广泛的分子注释,而应区分预测性、药效动力学和耐药性生物标志物。需要捕捉 XPO1 依赖性的分子特征,可能包括 NPM1、DEK::NUP214、SF3B1 和其他病变,以及靶点参与的药代动力学和药效动力学标志物,以预测疗效和耐受性,并支持个体化给药和患者选择。
总体而言,当前数据支持 XPO1 作为 AML 中一个生物学上引人注目且临床上充满希望的靶点,但广泛采用尚需更强有力的证据。XPO1 抑制剂不太可能作为所有复发/难治性病例的通用解决方案,但也不应被简单地视为仅具有毒性或边缘性的选择。关键的临床问题在于,在明确定义的亚组和治疗环境中,适当剂量和合理联合的 XPO1 抑制剂是否能在将增加的毒性和经济负担控制在可接受范围内的同时,有意义地提高再次缓解的概率、增加异基因移植的机会或延缓高危克隆的扩增。回答这些问题需要基于分子分型的大型、多中心、随机试验,并辅以长期随访和真实世界数据。只有通过这种综合努力,才能确定 XPO1 靶向策略在不断发展的 AML 治疗格局中的精确位置。
参考文献
Liu Y, Yun X, Ding W, Li S, Liu H. Targeting the nuclear export receptor exportin-1 in acute myeloid leukaemia: From biology to clinical translation. Clin Transl Med. 2026;16:e70676. https://doi.org/10.1002/ctm2.70676

