【BCD】T-ALL的生物学和遗传学基础及基于基因组的风险分层
时间:2026-05-08 20:22:44 热度:37.1℃ 作者:网络
深度解析医学证据,lxfs.net为你支撑决策
复发/难治性T细胞急性淋巴细胞白血病(T-ALL)预后极差,因此迫切需要识别高危患者。T-ALL的分型历来依赖于免疫表型,包括早期T细胞前体ALL的识别,但很少有基因改变能够独立于可测量残留病而可重复地预测T-ALL的结局。
《BLOOD CANCER DISCOVERY》近日发表文章,对 T-ALL 的生物学和遗传学基础进行了综述,重点是大规模的基因组分析,揭示了基因亚型的谱、其驱动因素、癌基因激活机制和发育阶段。文章还描述了基于基因组学的方法如何改善 T-ALL 分类和风险分层。

引言
更深入地了解ALL的生物学机制,可以通过更精确的风险分层和个体化治疗策略,提高儿童和年轻成人ALL的治愈率。ALL的风险分层整合了生物标志物与人口学、临床和治疗反应因素。理想情况下,风险适应性治疗将引导高危复发或难治性疾病患者接受强化或替代治疗,而低危患者则可受益于标准化疗或考虑降级治疗以最小化治疗相关毒性。然而目前唯一在多个T-ALL队列中进行过测试的基因组风险分类器是一个五基因分类器,且在不同队列中的表现不一。由于以下几个挑战,鉴定可重复预测预后的基因组特征的进展有限。首先,虽然小型研究已鉴定出非编码区的改变,但很少有病例进行过WGS,可能限制了生物学亚型和驱动因素的发现。其次,尚无既往研究有足够的能力来完全检测与MRD无关的预后相关改变。第三,既往一些研究中排除了诱导失败病例,可能掩盖了难治性疾病的驱动因素。最后,尽管通过免疫表型定义的ETP ALL被归类为高危并纳入T-ALL的风险分层,但该亚型的基因组驱动因素尚未完全明确,其预后意义在不同的临床研究中存在差异。基于基因表达的分子亚型历来仅得到部分解析,且临床效用有限,部分原因是缺乏全面的WGS分析和临床有用的基因组生物标志物。数据表明,基因组改变和分子亚型可与临床变量(包括MRD、外周血WBC计数和诊断时CNS受累情况)一起用于对T-ALL患者进行准确的风险分层。
本综述总结了T-ALL分子和遗传学分型的进展,强调了生物学信息分类框架如何作为改善T-ALL预后分层和患者结局的基础。
临床预后特征
MRD、诊断时WBC计数和CNS受累程度是仅有的、在不同研究组使用的不同治疗方案中始终显示具有预后意义的临床特征,在纳入体细胞基因组改变的多变量模型中也保持了预后意义。这些变量对结局的影响因试验和队列而异,如前所述。简而言之,MRD是T-ALL治疗反应和长期预后最可靠的生物标志物。不同的联盟使用不同的平台来量化MRD,包括流式细胞术、NGS和基于PCR的检测。大多数方案强调两个核心时间点:诱导治疗结束时(约为诊断后1个月)和巩固治疗结束时(约为诊断后3个月)。
诊断时的WBC计数是另一个常用的预后特征。虽然在B-ALL中使用50,000/μL的截断值进行风险分层,但WBC在T-ALL中的预后影响具有更高的异质性。值得注意的是,在COG AALL0434研究中,极高的WBC计数与非ETP T-ALL患儿的较差生存率相关。
诊断时的CNS状态也在风险评估中发挥作用。根据脑脊液细胞离心涂片和WBC计数,将患者分为三类:CNS-1(未检出原始细胞)、CNS-2(原始细胞阳性且CSF WBC <5/μL)和CNS-3(原始细胞阳性且CSF WBC ≥5/μL)。在T-ALL CNS-3患者中,尽管接受了强化的CNS定向治疗和颅脑放疗,但结局较差。
T-ALL的当前治疗
T-ALL接受约2至2.5年的多药化疗,通常包括6至9个月的强化多阶段治疗,随后是长期维持治疗。与B-ALL相比,T-ALL方案通常更强,且统一包括含蒽环类药物的诱导治疗。CNS导向治疗是所有方案的核心要素,使用反复的鞘内化疗和具有CNS活性的药物。国际协作组在几个临床相关组成部分上存在差异,包括是否完全省略颅脑放疗或仅保留用于特定患者(如诊断时CNS疾病负荷高的患者)。不同方案在鞘内治疗的强度和组成上也各不相同,一些方案对所有患者仅使用鞘内甲氨蝶呤,而其他方案则对CNS受累患者使用鞘内三联疗法。奈拉滨(nelarabine)的使用是另一个主要分歧点,一些工作组在一线治疗中广泛使用,而其他组则根据可及性、费用和风险分层将其限制用于高危或难治性疾病。总的来说,成人方案保留相似的主体药物,但通常会降低强度并修改对毒性敏感的组分(如皮质类固醇、门冬酰胺酶和CNS导向治疗要素),而儿童方案剂量强度更高,且在青少年和年轻成人中结局更好。首次完全缓解后分配至HSCT在各组间存在异质性,通常仅用于极高危患者或早期反应不充分的患者,但不同协作组之间的阈值和实践模式不同。最后,罕见的BCR::ABL1病例可使用激酶抑制剂靶向治疗。
T-ALL的免疫表型亚型
基于流式细胞术的免疫表型可用于T-ALL分类。EGIL定义了T-ALL的四种亚型:祖T细胞型(pro-T)、前T细胞(pre-T)/未成熟型、皮质型和成熟T-ALL型。这些免疫表型亚型反映了经典T细胞发育的关键阶段,其中TCRδ、TCRγ、TCRβ或TCRα位点的V、D和J基因片段体细胞重组决定了向表达γδ或αβ TCR的T细胞谱系的分化。Pro-T和pre-T/未成熟ALL对应于完全αβ或γδ重排前的早期胸腺细胞阶段;皮质ALL与αβ重排进行中的CD4+CD8+双阳性阶段一致;成熟ALL则反映了具有完全重排αβ TCR的单阳性CD4+或CD8+阶段。虽然皮质期β或成熟αβ TCR重排的T-ALL最常见,但一部分T-ALL遵循γδ T细胞谱系轨迹表达γδ TCR。
ETP ALL代表一种生物学上独特的白血病亚型,最初于2009年描述。其免疫表型定义为存在祖T细胞标志物、缺乏CD1a和CD8、CD5表达降低或缺失、以及共表达一个或多个干细胞或髓系标志物。这一特征表明发育阻滞在早期造血祖细胞阶段。一个相关的亚组称为“near-ETP” T-ALL,表现出相似的标志物谱,但保留了高CD5表达。
WHO和ICC的T-ALL分类系统通过免疫表型将ETP ALL与其他T-ALL区分开来。ICC分为三个主要亚型:ETP ALL、BCL11B激活型T-ALL和NOS T-ALL,并包含八个临时基因组亚组。因此,ICC将ETP免疫表型类别与额外的基因组亚型相结合,而WHO分类尚未将T-ALL的基因组亚型纳入分类,因为其临床意义仍不确定。
基于免疫表型的分类提供了对T-ALL与正常T细胞分化之间关系的见解,但其临床效用仍然有限。虽然一些研究报告了基于EGIL分类的生存差异(单变量分析中皮质T-ALL结局最好),但其预后意义在多变量模型中根据治疗反应进行调整后减弱。最初,ETP ALL在接受St. Jude和AIEOP方案治疗的患者中与不良预后相关。事实上,ETP ALL患者更可能经历原发性难治性疾病,而非ETP T-ALL患者在初始缓解后更可能复发。然而最近的研究表明,ETP ALL患者的生存率与非ETP T-ALL患者相当,突显了免疫表型ETP分类作为基因组风险替代指标的局限性。这些相当的生存结局可能部分反映了治疗差异。ETP ALL更常与皮质类固醇耐药和较差的早期MRD反应相关,因此ETP ALL患者通常被分层到更高危的治疗组并接受强化方案。另一个可能解释相当生存率的原因是,通过免疫表型定义的ETP ALL在生物学上并不精确,它聚集了高危和低危的基因组亚型。
T-ALL中基于免疫表型的分类效用有限的一个合理解释在于,白血病原始细胞通常表现出异质性和异常抗原表达,这可能仅部分重现了EGIL类方案所基于的正常T细胞成熟轨迹。此外,在某些病例中,谱系可塑性使白血病细胞能够获得髓系甚至B系标志物,模糊了类别界限。例如,BCL11B改变亚型根据免疫表型曾被归类为ETP ALL、T-ALL、AML或T/髓系MPAL。然而基因组分析揭示,这些病例的特征危BCL11B激活改变,并表现出相似的基因表达谱,从而强调了它们共同的生物学特性。同样,ETP ALL分类依赖一组有限的标志物将T-ALL分为三个免疫表型类别。大规模基因组分析表明,具有相似免疫表型的T-ALL病例表现出高度多样化的突变谱和不同的转录亚型。这凸显了基于免疫表型的T-ALL分类的局限性,其缺乏捕捉疾病潜在分子异质性所需的分辨率。
非编码改变作为T-ALL中癌基因激活的主要驱动因素
T-ALL主要由调控T系或HSPC程序的转录因子(TF)的表达失调驱动,或者较少见地由非HSPC或T系特异性的TF驱动。一个定义性特征是增强子劫持,即TCR和BCL11B位点的T系特异性元件异位激活癌基因;白血病细胞也利用T系信号通路(尤其是NOTCH1)来维持转化。尽管非编码改变被认为是T-ALL白血病发生的核心,但它们仍然难以检测,导致在驱动基因目录中系统性代表性不足。
近60%的 T-ALL 亚型定义改变位于非编码区,强调了 WGS 在大型基因组队列中检测这些事件的关键作用。涉及MYC、NOTCH1、ZNF219和 IRX3 的非编码改变也经常作为继发性共病变,共同存在于12.7%的病例中,然而亚型定义的非编码病变更危普遍。非编码改变主要通过促进增强子劫持的染色体重排或直接修饰调控元件来驱动癌基因激活。例如,T系增强子(如TCR和BCL11B相邻的ThymoD增强子)的劫持在44.5%的病例中观察到,并且是多个T-ALL亚型的标志。类似地,调控元件改变在TAL1 DP/αβ样亚型中常见,在24.7%的病例中检测到改变。T-ALL中癌基因失调控的另一个机制涉及破坏mRNA剪接和改变蛋白质结构的基因内重排(图1)。一个显著的例子是NOTCH1,其中内含子SNV、基因内缺失或内部串联重复修饰了胞外结构域,导致异常的NOTCH1激活。在这项研究中,双端WTS和长读长亚型测序有助于评估非编码变异对RNA剪接的影响,提供了对其功能后果更深入的了解。其他非编码机制包括NOTCH-MYC增强子扩增、14q32 BCL11B增强子串联扩增以及通过局灶性缺失实现的IRX3启动子栓系。还描述了激活致癌回路的非基因机制。许多非编码机制的一个关键特征是它们能够劫持T系特异性增强子元件,利用T系的转录机制来激活癌基因。另一个共同主题是新生增强子、新生启动子或启动子劫持的产生,所有这些都导致异常的TF结合和癌基因激活,如 MYB 结合位点生成导致 TAL1 激活。
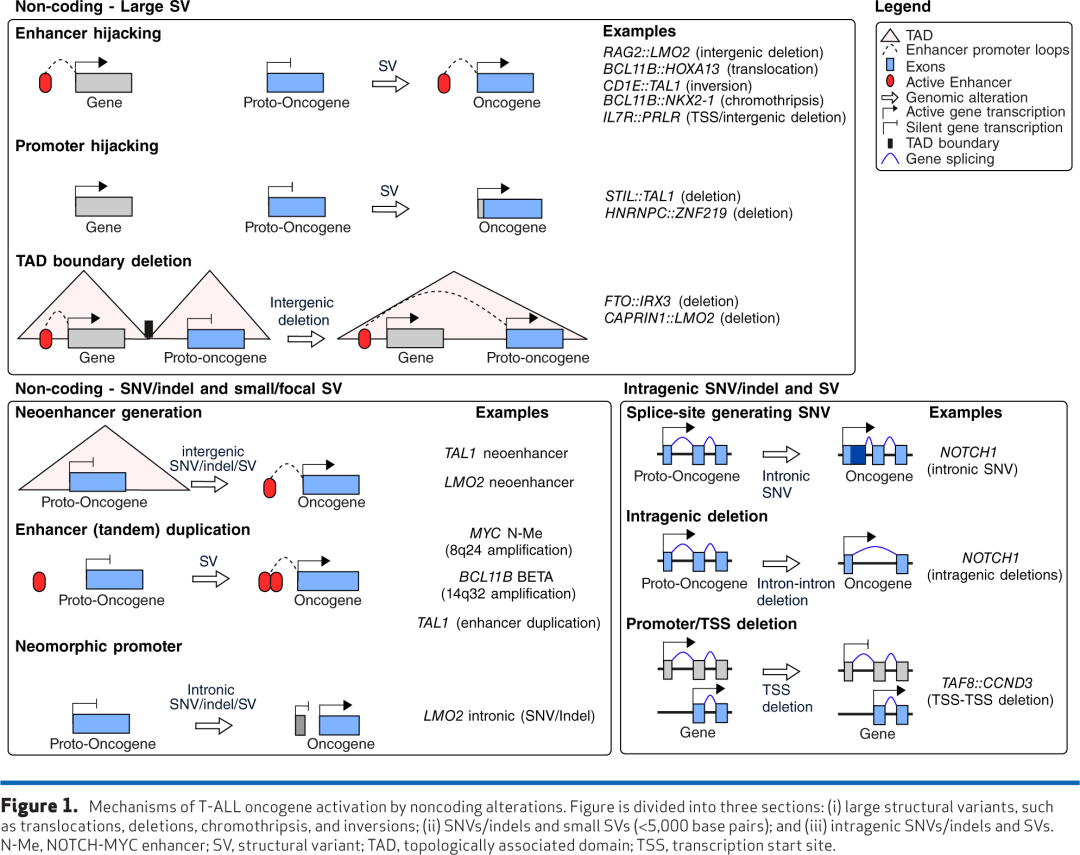
T-ALL中非编码改变的产生机制尚未完全阐明,然而有证据表明RAG活性可能产生这些改变的一部分。在T细胞发育的双阳性阶段,RAG表达高并促进TCRβ重排,使该阶段特别容易受到异常的RAG介导的对癌基因和TCR增强子附近隐秘重组位点的识别。这可能解释了阻滞于DP样阶段的亚型与TCR增强子劫持之间的强关联,后者是T-ALL中最常见的增强子劫持事件。相比之下,BCL11B增强子在更宽的发育窗口期(从ETP阶段到成熟T细胞)都具有活性。因此,这些增强子可用于被各种癌基因在多个T-ALL亚型中劫持。串联扩增侧翼有自身互补同源序列,可能在复制过程中促进二级结构的形成。
整合基因组和转录组测序能够稳健地发现非编码驱动因素。WGS检测劫持T系增强子以激活亚型特异性TF的结构变异,而WTS将断点与异位启动子使用和等位基因特异性表达联系起来。染色质和三维基因组检测(包括ATAC测序、Hi-C、HiChIP、CTCF ChIP-seq和H3K27ac分析)通过建立增强子可及性、活性、增强子-启动子接触和TAD边界,验证了增强子劫持并定义了染色质结构。
T-ALL的基因组和分子亚型
基因表达谱分析在揭示T-ALL中关键T-ALL致癌驱动因素和常见分子亚型方面发挥了关键作用,最常见的T-ALL分子亚型,以前称为TAL1-RA和TAL1-RB,由TAL1/TAL2/LMO1/LMO2/LYL1失调定义。其他基于基因表达的亚型包括NKX2-1、TLX1、TLX3和HOXA9激活亚型。通过重点基因组研究发现了额外的罕见亚型,包括SPI1重排、HOXA13激活、BCL11B激活和STAG2/LMO2相关T-ALL。
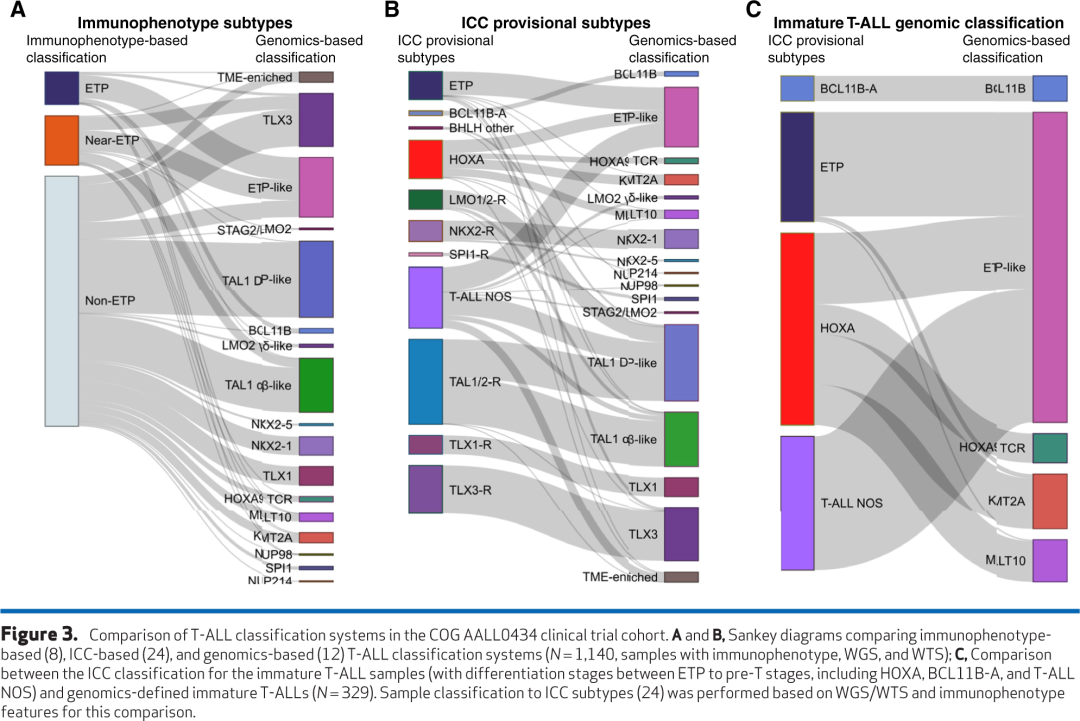
TARGET项目通过WES、WTS和SNP芯片分析了来自AALL0434临床试验的264例T-ALL病例。该研究确定了8种分子亚型和10个经常改变的通路中的106个推定驱动基因。然而,由于排除了经历IF的患者、队列规模相对较小以及大多数患者缺乏WGS,该研究未能识别所有驱动因素,从而限制了开发包含基因组改变的稳健预后模型。这些局限性促使由COG、费城儿童医院和St. Jude儿童研究医院的研究人员组成的联盟,在Gabriella Miller Kids First计划的支持下,对完整的AALL0434队列进行了基因组、外显子组和转录组测序。这项工作根据亚型定义性驱动因素、协同改变、基因表达和发育阻滞阶段确定了15种分子亚型(表1;图2)。T-ALL亚型与临床结局显示出强关联,凸显了它们作为预后生物标志物的潜力。基因组定义的亚型提供了对所有基于免疫表型的亚型和几个ICC临时亚型的精细重分类(图3A-C)。
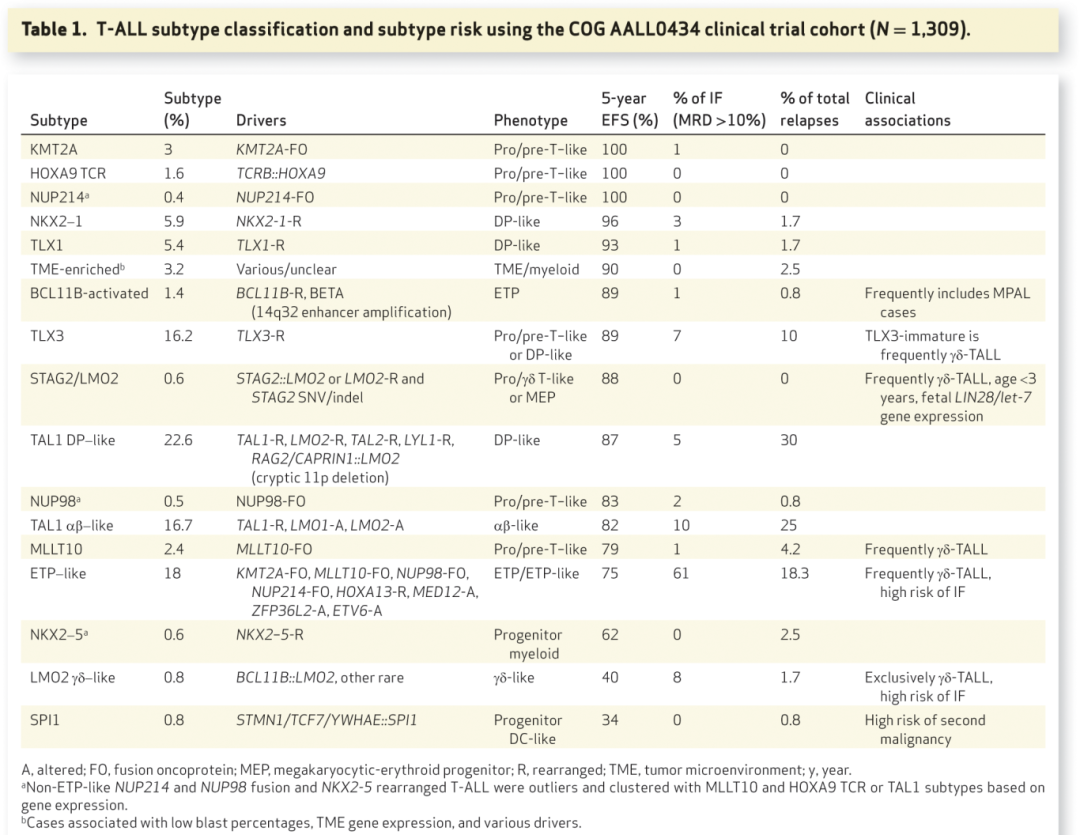
TLX1、TLX3和NKX2-1亚型
TLX1和TLX3是孤儿同源盒蛋白,其表达与胚胎发育有关。尽管TLX1和TLX3在造血系统中不表达,但这些基因被异常表达并经常发生重排:TLX1激活通过与TCRβ或TCRδ位点的重排实现,但也通过LINC00592和TLX1之间与TLX1激活相关的复发性基因间缺失实现。TLX3亚型的标志是通过BCL11B或TCR增强子劫持实现TLX3激活,伴有高频率的黏连蛋白通路改变。基因表达谱分析揭示了两个不同的亚组:“TLX3-未成熟”表现出更高频率的WT1、16q22.1/CTCF和DNM2改变,以及高频率的NUP214::ABL1融合和JAK-STAT改变。“TLX3 DP样”分化程度更高,具有高CD4和CD8表达,以及更高频率的LEF1/MYB/14q改变。因此,TLX3激活跨越了一个广泛的发育窗口(图2),其中阶段和协同改变可能导致不同的疾病生物学和临床特征。例如,TLX3-未成熟已被证明预后比TLX3 DP样更差,并且与多种失调激酶信号的共病变相关。
NKX2-1是T-ALL中一个经常发生重排的TF,在正常T细胞分化过程中不表达。它在胚胎发育过程中的肺和甲状腺器官发生和细胞分化中起关键作用。此外,NKX2-1在肺癌和甲状腺癌中经常被扩增/突变。根据基因表达和不同的NKX2-1激活机制,将NKX2-1亚型分为两个遗传亚组:“NKX2-1 TCR”表现出TCRδ劫持事件和RPL10突变,而“NKX2-1其他”以其他激活机制为特征,包括导致BCL11B增强子劫持的14号染色体重排以及NFKBIA增强子劫持的病例。NKX2-1值得注意的共病变包括RB1、LEF1、XPO1和通过HNRNPC启动子劫持激活ZNF219。TLX1、TLX3和NKX2-1亚型的一个概念性独特特征是,在pre-T/DP阶段,这些非T系TF被T系特异性增强子异位激活,导致异常的基因表达程序,从而在这些亚型中促进白血病发生。
TAL1亚型及TAL1/TAL2/LMO1/LMO2/LYL1调控网络的失调
bHLH TF TAL1、TAL2和LYL1的遗传改变(它们编码造血和T细胞发育的关键调节因子)是互斥的,并作为T-ALL TAL1亚型中定义亚型的致癌事件。此外,LMO1和LMO2改变也是常见的TAL1亚型驱动因素,彼此互斥,但经常与TAL1、TAL2和LYL1改变同时发生。LMO1和LMO2是LIM结构域蛋白,编码介导蛋白质-蛋白质相互作用和形成多蛋白造血转录复合物的转录辅助调节因子。值得注意的是,尽管LYL1和LMO2改变仅在TAL1/LMO2亚型中常见,但这些基因在多个祖细胞T-ALL亚型中过表达,包括BCL11B和ETP样T-ALL亚型,凸显了它们在T-ALL白血病发生中更广泛的作用。
根据分化阶段,TAL1亚型分类为αβ样和DP样亚型。这些亚型中的已知驱动因素包括STIL::TAL1(通过STIL启动子劫持激活TAL1),以及激活TAL1、TAL2、LMO1、LMO2和LYL1的TCRδ或TCRβ增强子劫持重排。此外,还观察到非编码序列突变导致的TAL1激活,包括通过小插入形成TAL1新增强子、TAL1内含子SNV、TAL1下游增强子上的小拷贝数增益、LMO1增强子突变,以及通过SNV/indel和小拷贝数增益产生LMO2新形态启动子或新增强子。尽管这些亚型的驱动改变多种多样,但TAL1 αβ样更常与STIL::TAL1和LMO1增强子SNV相关,而TAL1 DP样与LMO2、TAL1和LYL1的TCR劫持相关。其他复发性病变包括导致CAPRIN1::LMO2的11p基因间缺失,以及TAL1 DP样特异性的RAG2::LMO2增强子劫持事件。TAL1 DP样亚型具有高表达CD8、CD4、RAG1和RAG2的特点,反映了DP胸腺细胞阶段的分化阻滞。相比之下,TAL1 αβ样亚型具有高表达TRAC基因和经常出现TCRα重排的特点。发育阶段的差异可能影响这两种亚型中驱动激活机制和协同病变的谱系。
在TAL1亚型中观察到几个共发生病变和通路改变表现出互斥性,包括RPL10突变、6q缺失以及LEF1、JAK-STAT、PI3K和NOTCH通路的异常。这些互斥模式通常富集在不同的转录组亚组中,表明遗传改变和基因表达谱共同有助于定义TAL1驱动的T-ALL中生物学和临床相关的亚组。TAL1 DP样亚组进一步分为:RPL10突变的病例;携带LEF1缺失或结构变异或LYL1改变的病例;JAK-STAT通路基因突变的病例;以及具有其他遗传改变的多样性组。TAL1 αβ样亚型进一步分为三个遗传类别:伴有PTEN缺失和PI3K通路突变但无NOTCH1突变的病例;涉及6q缺失的病例;以及具有其他改变(如PI3K通路突变和NOTCH1突变)的异质性组。关键的概念性发现是,TAL1亚型的特征不是单一的驱动改变,而是分化阶段以及TAL1/TAL2/LYL1和/或LMO1/LMO2转录网络的失调。此外,驱动改变表现出亚型差异,如RAG2::LMO2和TAL1 DP样中TCR劫持的发生率更高。这可能与DP分化阶段有关,在该阶段RAG2具有活性,细胞经历TCRα重排。此外,共病变和驱动因素可能通过协同相互作用和基因调控依赖性相互关联。例如,产生新增强子的改变可通过创建MYB结合位点来激活TAL1表达,而NOTCH1激活则通过N-Me增强子在多个亚型中促进MYC表达。MYC表达在TAL1 DP样亚型中经常升高,这可能是由于FBXW7突变发生率更高,已知这会导致更高的MYC蛋白水平。相比之下,TAL1 αβ样亚型表现出MYB和MYCN表达增加,而LMO2 γδ样和STAG2/LMO2亚型分别以MYCN和MYC表达为特征。MYCN突变富集在TAL1 αβ样和LMO2 γδ样中,但在TAL1 DP样亚型中没有,进一步凸显了癌基因表达、主要驱动因素和共发生病变之间复杂的相互依赖性,所有这些都受到白血病分化阶段的塑造和约束。
BCL11B激活型白血病
BCL11B激活型亚型是一种祖细胞来源的白血病,通过免疫表型先前被分类为T/髓系MPAL、ETP ALL、AML。该亚型由BCL11B激活驱动,激活机制为:通过染色体间重排劫持通常在HSPC中具有活性的增强子,从而驱动BCL11B的阶段不适当失调表达;或通过14q32上BCL11B下游非编码区的局灶性扩增,产生激活BCL11B的新增强子。BCL11B激活型亚型高度富集FLT3 ITD突变,这在其他ETP样ALL病例中很少见。研究表明,BCL11B激活型亚型在MPAL、ETP ALL和AML免疫表型分类中表现出一致的基因表达谱。鉴于共享的驱动改变,这些病例应被视为一个独特的白血病亚型,这也反映在当前的ICC分类中。需要注意的是,由遗传改变导致的BCL11B激活是BCL11B激活型亚型所独有的,因为成熟T-ALL亚型更常与BCL11B功能丧失突变和BCL11B增强子的劫持相关。
ETP样ALL及其与既往定义的T-ALL亚型的区别
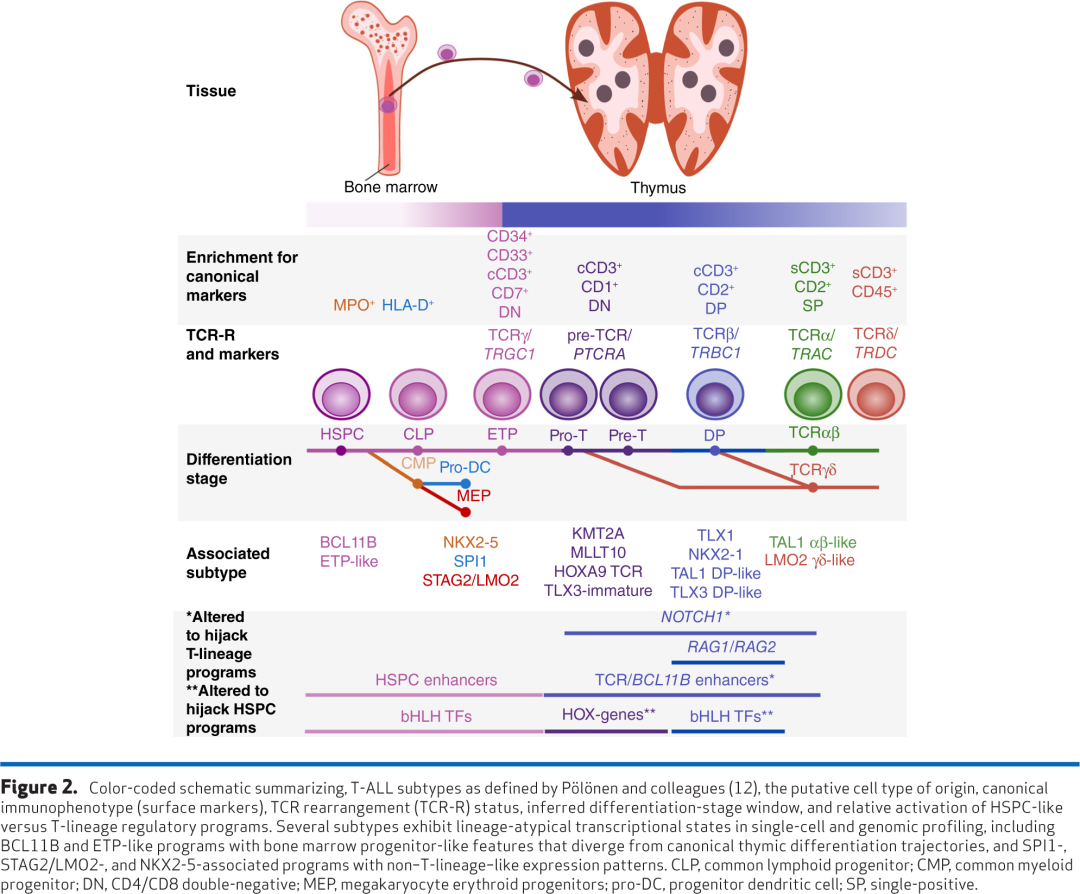
Pölönen及其同事报告的一个关键发现是,具有ETP或near-ETP免疫表型的病例分散在T-ALL的转录谱和基因组亚型中;相反,识别出富集ETP ALL的病例簇,这些病例簇也包含非ETP ALL病例。具体来说,在TAL1和TLX3重排以及BCL11B激活型白血病的一个子集中观察到ETP ALL免疫表型病例,但最常见于一个称为“ETP样”T-ALL的独特基因表达定义的亚型中。尽管大多数ETP-IP病例聚集在ETP样基因组组内,但该组也包含相当比例的near-ETP病例和较小比例的非ETP病例。相反,在ETP样亚型中,38%的病例被归类为ETP,34%为near-ETP,28%为非ETP。值得注意的是,在ETP样基因组组中,驱动改变的频率在ETP、near-ETP和非ETP免疫表型病例中是相似的。此外,不同免疫表型的病例在ETP样组内未基于基因表达形成不同的簇(图4A)。
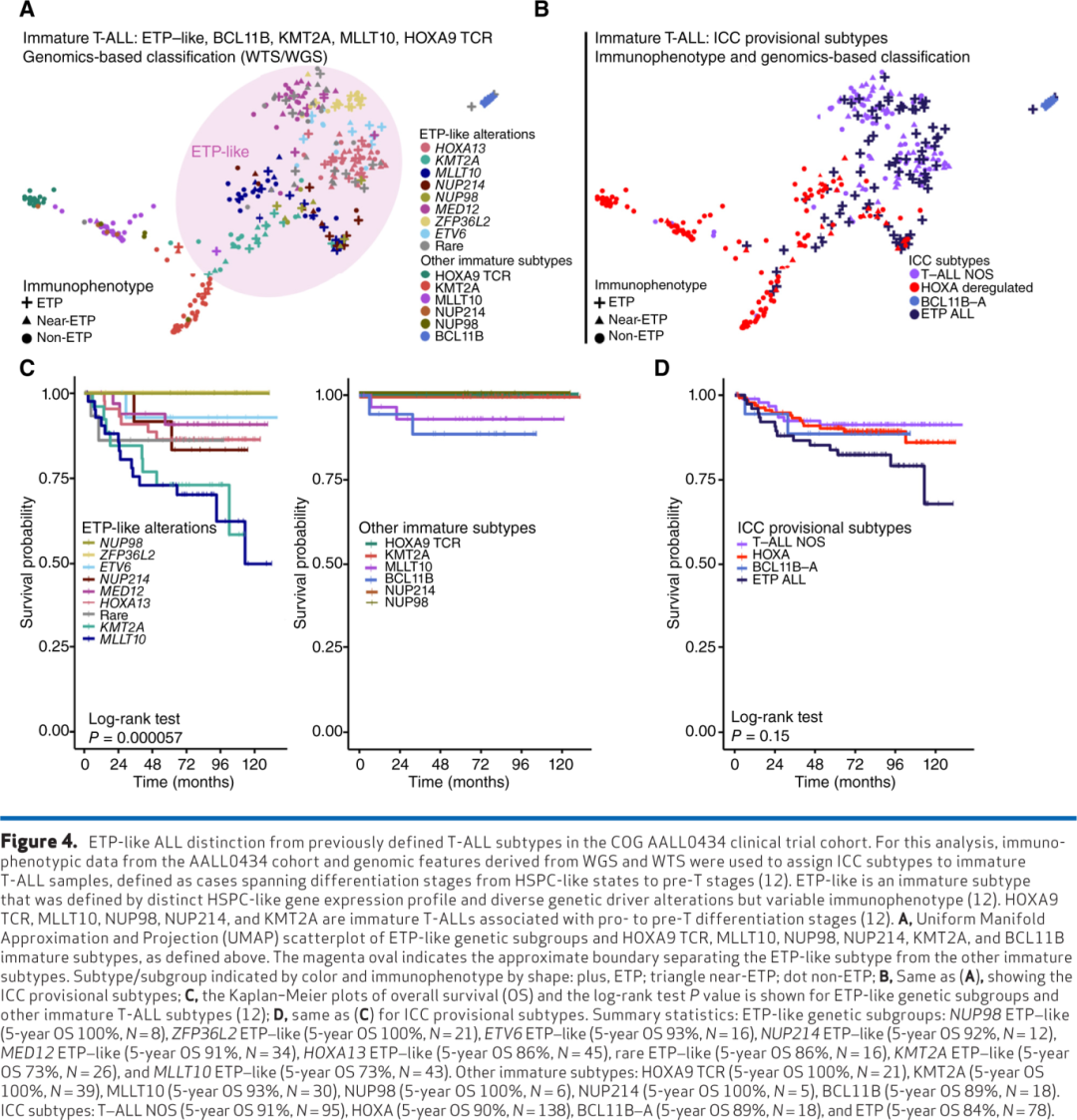
ETP样亚型由编码已知或潜在造血干细胞功能调节因子的基因中的多种驱动改变所定义。这些包括涉及MLLT10、KMT2A、NUP98和NUP214::SET的HOXA9激活型融合癌蛋白;HOXA13激活型重排;以及ZFP36L2、ETV6、RUNX1和MED12中的复发性改变。值得注意的是,ETP样基因组亚型通过基因表达与更分化的亚型(也包括一些相同的改变靶点:MLLT10和KMT2A重排以及TCRB::HOXA9 T-ALL)分开聚类。HOXA9激活型病例以前被归在HOXA失调T-ALL的总称下。为了将ICC-未成熟T-ALL类别与已发表的基于基因组的分类进行比较,本文根据WTS/WGS数据将AALL0434队列分类为ICC类别。尽管在ETP样组中也观察到涉及KMT2A或MLLT10的重排和升高的HOXA9表达,但它们伴随着与ETP样组外观察到的病例不同的转录程序、发育阻滞点、共突变谱和临床结局(图4A和C)。KMT2A亚型主要由KMT2A::MLLT1融合癌蛋白驱动,而ETP样KMT2A病例主要与KMT2A::AFDN融合相关。HOXA9 TCR亚型与ETP样白血病相比表现出更分化的状态。值得注意的是,TCRB::HOXA9重排断点仅与HOXA9激活相关,而HOXA13激活是ETP样亚型特有的,并与不同的断点位置相关。比较分析表明,ICC-未成熟T-ALL亚型与结局无关,而基于基因组的类别可能具有预后意义,尽管需要更多研究来进一步支持跨亚型的预后潜力(图4C和D)。值得注意的是,早期的基因表达微阵列研究定义了一个与ETP样重叠但也包括BCL11B/KMT2A亚型的“未成熟”程序。总之,驱动改变、独特的基因表达谱和分化阶段将ETP样ALL与先前定义的未成熟、BCL11B激活和HOXA失调亚型以及免疫表型定义的ETP ALL区分开来。
LMO2 γδ样亚型
LMO2 γδ样亚型的定义是独特的基因表达谱、成熟的γδ T细胞表达特征和γδ TCR重排。尽管遗传上多样,但LMO2 γδ样亚型通常由复发性改变驱动,如BCL11B::LMO2和TCRD::MYC重排,以及MYCN突变和LMO2调控元件中的SNV或indel。
STAG2/LMO2亚型
STAG2功能丧失突变已在多种癌症中观察到,包括AML。STAG2是黏连蛋白复合物的核心组分,对维持正确的3D基因组结构、促进增强子-启动子相互作用以及确保发育和分化过程中的准确基因表达程序至关重要。在T-ALL中,STAG2/LMO2亚型表现出独特的基因表达谱和靶向LMO2和STAG2的双重打击改变。T-ALL中最常见的改变是STAG2::LMO2重排,它通过劫持STAG2增强子同时使STAG2失活并激活LMO2。其他机制包括STAG2的功能丧失突变和LMO2的其他激活改变。机制上,STAG2通过扰乱增强子-启动子相互作用破坏染色质结构,导致影响T细胞分化关键通路的广泛基因表达变化。此外,STAG2/LMO2亚型与LIN28/let-7通路的激活相关,该通路通常在胎儿HSPC中表达,出生后沉默。这表明STAG2/LMO2亚型可能起源于胎儿HSPC。
SPI1亚型
SPI1/PU.1是一个在造血分化和谱系决定中起关键作用的TF,特别是在髓系和淋巴样细胞中。SPI1融合癌蛋白,包括TCF7::SPI1、STMN1::SPI1和YWHAE::SPI1,以及独特的基因表达和甲基化谱是SPI1亚型的标志。该亚型与髓系/树突状细胞祖细胞基因表达特征以及HLA-DR和sCD3的表达相关。
其他罕见或模糊亚型
其他亚型包括与ETP样NUP214::SET和NUP98病例分开聚类的NUP214::SET和NUP98的离群病例,表明分化阶段更成熟。此外,NKX2-5激活是一种罕见亚型,通常具有来自其他亚型的离群病例和髓系基因表达。还识别出一个具有高TME参与、低原始细胞百分比和多种驱动因素(称为“TME富集”)的亚型。较低的纯度可能是驱动独特的基因表达聚类的原因,而不是这些病例的生物学差异。
非经典分化轨迹与γδ T-ALL基因组学
表达γδ TCR的T细胞是正常造血中的一个罕见亚群,约占外周T细胞的0.5%至5%,代表了T细胞发育中的一种替代分化路径。γδ TCR重排的T-ALL约占T-ALL病例的10%,直到最近在基因组水平上仍知之甚少。Kimura及其同事分析了200例儿童γδ T-ALL病例,并将数据与AALL0434队列进行比较,揭示了与非γδ T-ALL相比不同的基因组亚型分布。γδ T-ALL病例以亚型依赖的方式基于基因表达与非γδ T-ALL病例聚集在一起,并共享几个相同的驱动改变,但驱动重排的频率差异很大,反映了几个失调/重排TF具有活性的分化阶段。几个亚型,如TLX3-未成熟、STAG2/LMO2、KMT2A、MLLT10和ETP样,同时包含γδ T-ALL和非γδ T-ALL病例,而TAL1、TLX1和BCL11B在γδ T-ALL中不存在。LMO2 γδ样亚型是唯一一个根据TCR重排完全由γδ T-ALL病例组成的亚型,并表现出γδ T细胞基因表达特征。这些研究的另一个关键发现是,TCR重排状态(无论是缺失还是涉及β、γ、αβ或γδ重排)反映了转化时的成熟阶段。此外,标志物基因表达和TCR重排表明,大多数T-ALL在达到效应T细胞成熟阶段之前就发生了转化。
成人和婴儿T-ALL
尽管成人和婴儿T-ALL与儿童和年轻成人疾病共享相同的总体分子亚型,但它们的亚型组成不同。在婴幼儿中,未观察到BCL11B、HOXA9 TCR、TLX1和TLX3亚型,而STAG2/LMO2、SPI1和NKX2-1或NKX2-4或NKX2-5亚型显著富集,占病例的27%,而在儿童和年轻成人队列中低于10%。婴儿和儿童组之间其他亚型的频率大致相当。在成人和年轻成人中,BCL11B、ETP样和HOXA TCR亚型发生更频繁。由于成人数据有限,尚未对完全通过WGS和WTS表征的成人和儿童队列进行全面的比较。成人队列始终显示更高的DNMT3A、IDH2、ASXL1和TET2突变率,但仍有相当一部分成人病例的基因组驱动因素未解明。
单细胞和表观遗传学分析的见解
单细胞分析已成为提高对T-ALL调控网络、治疗耐药机制和分化阶段理解的有力工具。Xu及其同事对40个T-ALL样本应用了单细胞RNA和ATAC测序,并将白血病原始细胞映射到人类T细胞发育轨迹上,识别出一种罕见的骨髓祖细胞样转录程序。该亚群在诊断时占<5%的原始细胞,表明该群体可能对治疗更具耐药性,从而对白血病风险分层产生更广泛的影响。支持这一跨谱系的概念,Chen及其同事报告说,T-ALL、AML和MPAL的一个子集共享一个与化疗耐药和不良结局相关的HSPC样群体,在ETP/ETP样病例中观察到最高频率。同时,Gower及其同事使用40个诊断样本的多模式单细胞分析,识别出在ETP样和TLX3未成熟亚型中富集的IRF5区域相关炎症特征。相反,一项将工程化人类T-ALL白血病发生模型与单细胞RNA-seq相结合的整合研究,定义了一个NOTCH1二聚化依赖的转录程序,包括HES4,该程序描绘了原发性T-ALL中不同的原始细胞亚群,并与不良免疫表型特征和较差结局相关。最后,Lim及其同事对58例儿童T-ALL病例进行的单细胞研究,识别出一个与IF病例相关的、具有先天样淋巴细胞特征的ZBTB16阳性非经典淋巴母细胞群体。这种可通过流式细胞术测量的标志物在诊断时存在,并预测较差的生存率。这些研究强调了将批量WGS/WTS基因组学和亚型与单细胞转录组分析相结合以解析失调通路的能力,为疾病生物学和治疗耐药机制提供了更深入的见解。
DNA甲基化分析已被用于多种恶性肿瘤的亚型分类和预后预测。肿瘤抑制基因启动子高甲基化、癌基因启动子低甲基化和整体甲基化模式已被证明与疾病表型、分化和遗传改变相关。DNA甲基化已成为分类T-ALL的有前途的生物标志物,有几项研究专注于定义全局表观遗传景观和基于甲基化的亚型。然而,在基于DNA甲基化的分类中,识别遗传驱动改变的差距构成了重大挑战。此外,需要对转录组/基因组亚型和DNA甲基化衍生的亚型进行系统性比较,以评估甲基化与基于WTS/WGS的分类方法之间的一致性。整合DNA甲基化、WGS和WTS的多组学分析有望完善T-ALL亚型、揭示癌基因失调的表观遗传机制,并识别新的预后生物标志物。这些是未来研究的重要方向,特别是因为异常DNA甲基化代表了去甲基化药物潜在的治疗靶点。
遗传和分子亚型的临床意义
ETP样、BCL11B和HOXA9激活驱动的亚型
ETP样T-ALL与难治性疾病(持续EOI MRD)和不良结局相关。重要的是,在ETP样亚型中,具有ETP、near-ETP和非ETP免疫表型的患者具有相似的基因组驱动因素和结局,凸显了基因组定义分类与基于免疫表型的分类相比的临床重要性。相反,BCL11B激活亚型具有与ETP样T-ALL不同的基因表达谱,尽管高度富集ETP免疫表型,但表现出相对有利的结局。总体而言,基于免疫表型的分类在预测结局方面表现不佳,无法区分具有不同预后影响的不同生物学组(图4C)。此外,数据支持用更广泛的、基因组定义的“ETP样”白血病亚型取代传统的ETP ALL免疫表型分类,后者更准确地反映了疾病的潜在生物学。尽管ETP样亚型中的大多数基因组亚组预后较差,但ETP样ZFP36L2亚组具有更高的MRD率,但与有利结局相关(图4C)。MLTL10重排与较差的结局和较差的MRD反应相关,特别是ETP样伴MLTL10重排的结局尤其糟糕。相比之下,ETP样亚型中的KMT2A重排病例具有较差的EOI MRD反应和较差的无事件生存期,而ETP样亚型外的KMT2A重排病例也具有较差的MRD反应但具有有利的EFS。当前的ICC和WHO对未成熟T-ALL的分类在预后上没有信息量,未能将ETP样与AALL0434队列中其他生物学上不同的组区分开来(图4)。O'Connor及其同事使用WGS分析了46例IF病例,识别出10个起始驱动事件,表明IF缺乏统一的遗传驱动因素。值得注意的是,该研究还发现大约一半的病例表现出ETP免疫表型。相比之下,对AALL0434队列数据的分析表明,基于基因表达的ETP样和LMO2 γδ样亚型占IF病例的69%,表明不良治疗反应很大程度上可以通过亚型特异性特征和潜在分化阶段来解释。
SPI1和组织细胞增生症
SPI1亚型在多项独立研究中一直与不良结局相关。SPI1亚型与第二癌症相关,特别是DC来源的朗格汉斯细胞组织细胞增生症和髓系肉瘤。值得注意的是,SPI1融合在第二癌症中得以保留,并伴有额外的克隆改变,例如BRAF突变克隆的扩增,提示从T-ALL到组织细胞增生症的组织病理学转化可能是一种谱系转换形式,而非真正的新发第二癌症。
LMO2 γδ样与难治性疾病
最近描述的LMO2 γδ样亚型与较差治疗反应和较差结局相关。LMO2 γδ样特征性的BCL11B::LMO2重排已在其他研究中观察到,并与IF一致相关。
TAL/LMO亚型和预后改变
TAL1亚型约占复发的55%,凸显了它们在疾病复发中的重要作用,而只有15%存在IF。在某些TAL1驱动改变中也观察到这种模式,如TAL1增强子indel和通过LMO2进行的RAG2/CAPRIN1增强子劫持,这些与更高的复发累积发生率相关。TAL1遗传亚组与结局有不同的关联,几个遗传亚组定义性改变是与MRD反应无关的预后因素。这反映在对各种TAL1遗传亚组定义性病变的单变量和复发累积发生率分析中;6q缺失、RPL10突变和NOTCH1突变是有利的,而NOTCH1野生型、TCRD::MYC重排以及TCR::LYL1和PTEN缺失与TAL1亚型内的不良结局相关。值得注意的是,与非亚型特异性改变(如PI3K通路改变和MYC增强子增益)与有利结局相关,相比之下,PTEN缺失和TCRD::MYC则不然。对于广泛突变的NOTCH1也出现了类似的模式;NOTCH1内含子SNV和基因内缺失以非亚型依赖的方式与较差的结局相关。这些结果凸显了需要进行WGS以全面检测和分类所有变异来评估预后意义。
STAG2/LMO2亚型主要在3岁以下儿童中观察到。尽管最近两项研究报告了该亚型的有利预后,但Pölönen及其同事、Kimura及其同事和Delafoy及其同事发现,年龄小于3岁、伴有STAG2::LMO2重排的γδ T-ALL患者结局极差。这些发现表明,γδ TCR重排状态可能影响STAG2::LMO2重排在T-ALL中的预后影响,但鉴于关于结局的结果存在分歧,由于该亚型的罕见性,这种关联需要在额外的独立队列中进行研究。
γδ T-ALL
γδ T-ALL与较差的结局相关。结局较差的一个原因可能是与不良预后相关的亚型(如MLLT10、LMO2 γδ样、TLX3-未成熟和ETP样)发生率更高。γδ T-ALL也具有不同的临床特征,表明γδ谱系定向可能影响免疫表型和临床特征。例如,γδ T-ALL患者通常具有成熟T免疫表型,其特征为年龄更小、诊断时WBC计数更高、无纵隔肿块,以及倾向于在骨髓而非CNS中复发。这些发现强调了使用TCR重排状态作为基因组生物标志物的必要性。
风险分层模型
基因组发现表明,将临床和基因组数据整合到多变量预后模型中可以增强T-ALL的风险分层。五基因分类器(NOTCH1、FBXW7、NRAS、KRAS、PTEN)最初在两个成人队列中研究,后来在儿童T-ALL队列中进行了测试(表1),但这些改变的预后价值一直不一致。NOTCH1和FBXW7野生型始终与较差结局相关,而这两个基因的突变通常与有利预后相关。相比之下,PTEN和KRAS/NRAS改变在UKALL2003队列中没有预测性,但在GRAALL-2003、GRAALL-2005和FRALLE2000T中具有预后意义。Simonin及其同事完善了四基因分类器,该分类器在成人和儿童患者中均显示出改进的预后分层。NGS panel包括72个经常突变的T-ALL基因的编码区,然后使用惩罚回归模型进行特征选择。作者随后开发了一个基于NGS的分类器,通过存在NOTCH1、FBXW7、PHF6和EP300突变来定义低危患者,通过PI3K通路基因(PTEN、PIK3CA、PIK3R1)、DNA甲基化相关基因(DNMT3A、IDH1、IDH2)、RAS通路基因(NRAS、KRAS)或其他基因(IKZF1和TP53)的突变来定义高危患者(图5A)。值得注意的是,尽管诊断时罕见,但TP53突变与未成熟表型、较差结局相关,并且经常在复发时获得。进一步的完善是将这个基于基因panel的分类器与WBC计数或通过PCR评估的EOI MRD整合,形成一个整合风险分层模型。作者旨在确定T-ALL中能够在临床环境中实现预后分类的最小基因组改变集。
DNA甲基化模式也显示出在完善T-ALL风险分层方面的潜力。CIMP的特征是广泛的DNA高甲基化,并与T-ALL中有利结局相关,而整体低甲基化与较差预后相关。Schäfer Hackenhaar及其同事应用EPIC v1.0和v2.0甲基化阵列将儿童T-ALL病例分为CIMP-high和CIMP-low组。当与EOI MRD状态结合时,CIMP分类具有独立于MRD的预后能力。在训练队列和验证队列中,低CIMP且MRD ≥0.1%的患者EFS显著更差,而高CIMP且MRD <0.1%的患者结局更有利。CIMP-low组富集了TAL1亚型特征,而CIMP-high组更常见HOX基因和TLX3表达。该研究的一个局限性是缺乏全面的分子亚型分类和WGS,这限制了对潜在基因组驱动因素的了解。
作者提出了新的风险分层方法,这些方法在设计上与以前的模型不同,因为它们基于WGS和WTS,并依赖疾病亚型和/或遗传改变作为预后生物标志物。提出了两个互补的风险分层框架来改善T-ALL的预后分类。首先,一个整合临床变量与五个分子亚型和18个基因组改变的惩罚Cox回归模型(pCox)(图5B、C)。重要的是,通过数据驱动的方式从所有显著突变的改变和T-ALL亚型中进行特征选择以获得预测特征。第二个模型是一个生存树模型,首先根据亚型或遗传亚组将患者分为风险组,然后根据MRD ≤0.1% vs >0.1%进一步区分风险组。pCox模型通过利用多个预后因素的加权贡献提供精确的风险分层,将患者分为大小相等的组,而ST模型允许使用与疾病分子亚型和遗传亚组一致的离散类别进行直观分类。
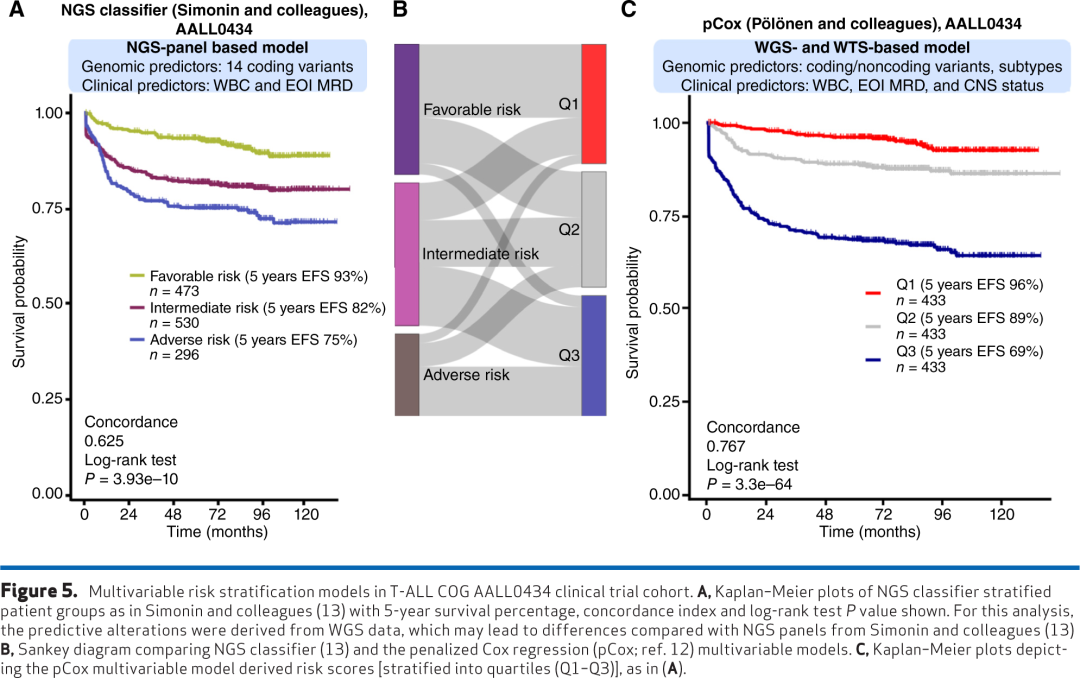
风险分层方法面临挑战,需要在跨越不同治疗、年龄和种族的独立队列中进行验证。Simonin及其同事的NGS分类器的一个关键限制是它没有考虑亚型信息或非编码改变,这些越来越被认为是T-ALL中的关键生物标志物。这一遗漏可能导致风险分层模型欠拟合。事实上,在AALL0434数据集中,尽管该模型仍然具有预后意义,但与pCox模型相比具有更差的一致性。这种方法的另一个限制是NGS panel不适合进行亚型分类。随着精准医学的进步,治疗策略可能会集中于靶向高风险亚型,如下文详述。尽管pCox和ST模型在训练数据和技术基准中显示出有希望的准确性,但这些模型也面临挑战。首先,它们需要WTS和WGS来推导预测特征,这对患者样本的数据生成提出了挑战。其次,用于诊断目的的变异检测存在计算挑战和需要基因组学专业知识。第三,这些模型是在儿童和年轻成人队列中拟合的,这可能在对成人队列进行分层时引入偏差。基于WTS和WGS的方法的一个关键优势是其适应性,这允许随着新基因组发现的出现,风险模型可以不断更新和基准测试。相比之下,靶向NGS panel需要重新设计和定制以纳入新发现。
靶向T-ALL
准确的T-ALL亚型分类有可能为亚型导向治疗策略的开发提供信息,但这需要前瞻性研究的验证。例如,高通量药物筛选已在伴有STAG2失活的T-ALL病例中(尤其是在STAG2/LMO2亚型内)识别出一个潜在的治疗脆弱性,表明这类白血病可能对靶向DNA修复通路的PARP抑制剂治疗敏感。
此外,由pre-TCR-LCK通路激活驱动的T-ALL病例可能对达沙替尼有反应,这一特征在TAL1、TAL2、LMO1和LMO2表达升高的白血病中尤为突出。其中,分别与单阳性αβ T细胞发育阶段和增殖性双阳性T细胞发育阶段相关的TAL1 αβ样和TAL1 DP样亚型,通常以高水平的LCK表达为特征。
由于TAL1亚型约占T-ALL的40%,使用达沙替尼治疗性抑制LCK是一种引人注目的治疗策略。此外,mTORC1抑制剂替西罗莫司已在异种移植模型中显示出与达沙替尼的协同作用,支持联合靶向通路。Toscan及其同事开发了一种新型前药ACHM-025,该药可被AKR1C3选择性激活,而AKR1C3在T-ALL中以亚型依赖性方式表达。该药物在T-ALL患者来源的异种移植模型中显示出强大的体内疗效。
ETP样和BCL11B亚型的特征是BCL2高表达,药物筛选已在BMP样/ETP样原始细胞中识别出一个治疗脆弱性,可通过维奈克拉有效靶向以诱导凋亡。此外,临床前研究表明,BCL11B亚型对使用维奈克拉和吉瑞替尼抑制BCL2和FLT3存在潜在的治疗脆弱性。使用芦可替尼靶向JAK/STAT通路也已在ETP ALL中显示出疗效,并且可能是伴有JAK/STAT突变的TAL1 DP样亚型的可行选择。
特定的激酶改变也可能是治疗靶点,包括PDGFRA改变、FLT3-ITD、可能还有NUP214::ABL1,以及在TLX3-未成熟和其他亚型中观察到的其他激酶融合或突变。然而,使用激酶抑制剂靶向NUP214::ABL1等继发性病变可能具有挑战性,因为这些改变通常是亚克隆的,可能限制反应的深度和持久性。因此,应在基因组解析的队列中评估治疗效果,并评估可靶向病变的克隆性。
最后,HOXA9激活型融合与高MEIS1表达相关,提示了一个潜在的治疗靶点。Menin抑制剂已在其他白血病中显示出疗效,可能为靶向与MEIS1表达相关的HOXA9激活型融合提供一种有前景的策略。
结论
基于基因组学的T-ALL分类与潜在的生物学更为一致,并提供具有临床意义的见解,而免疫表型亚型则缺乏同样水平的生物学粒度,临床效用有限。我们认为,整合全基因组测序和全转录组测序对于推进T-ALL研究越来越重要,这不仅是为了识别编码区和非编码区的驱动变异并提供亚型分类,也是为了潜在地改善风险分层。
将基因组发现转化为常规诊断和风险分层存在明确的未满足需求,在大多数情况下,这些诊断和风险分层仍然依赖传统的诊断工具。尽管基于基因组学的T-ALL分类目前尚未在临床中使用,且实施起来可能看似复杂,但它与B-ALL相似,而B-ALL的基因组分析现已在许多中心被应用于诊断。
未来研究的几个优先事项包括:(i) 在更多接受同质化治疗的大型儿童和成人T-ALL队列中,验证基因组驱动的分类、风险分层模型和预后生物标志物;(ii) 将这些框架转化到临床实践中,以改善患者结局;(iii) 开展亚型聚焦的机制研究,以理解复发/难治性疾病的机制,并为高危T-ALL亚型识别可靶向的基因;(iv) 为新发现的高危T-ALL亚型开发易于操作的细胞系和异种移植模型,以识别和验证治疗脆弱性。

