分子检测助力罕见原发性腹膜透明细胞癌诊断,检出dMMR+PIK3CA等突变,为个体化治疗提供新依据
时间:2026-05-08 20:03:33 热度:37.1℃ 作者:网络
原发性腹膜透明细胞癌(PPCCC)是一种极为罕见的恶性肿瘤,其临床和组织学特征与妇科上皮癌高度相似。目前对其发病机制知之甚少,可能起源于苗勒管化生或子宫内膜异位症的恶性转化。一名 48 岁女性,无子宫内膜异位症或激素治疗史,因急性下腹痛就诊。影像学检查发现盆腔肿块和胰尾部病变。患者接受了子宫切除加双侧输卵管卵巢切除术,以及远端胰腺切除加脾切除术。术中可见盆腔肿块位于腹膜,而妇科器官大体外观正常。对盆腔、胰腺和脾脏病变的组织病理学检查显示,三者均具有透明细胞癌的相同特征。免疫组化显示PAX8、CK7和HNF1β呈阳性,MSH2和MSH6表达缺失。下一代测序发现存在ARID1A缺失和体细胞PIK3CA突变。未检测到原发性卵巢、子宫内膜或肾脏肿瘤,支持原发性腹膜透明细胞癌伴胰腺和脾脏转移的诊断。本病例凸显了原发性腹膜透明细胞癌的诊断挑战,并为这种未被充分认识的恶性肿瘤的临床和病理提供了宝贵见解。
背 景
原发性腹膜透明细胞癌(PPCCC)是一种极为罕见的恶性肿瘤,在临床和组织学上均与妇科上皮癌高度相似。透明细胞癌(CCC)最常起源于卵巢或子宫内膜,而作为原发性肿瘤发生于腹膜的情况极为罕见。自 1990 年首次描述以来,英文文献中仅报道了有限的病例。由于PPCCC与更为常见的卵巢、子宫内膜和肾脏来源肿瘤的形态学特征存在重叠,因此诊断难度较大。目前PPCCC的发病机制仍不明确,现有假说认为其可能起源于苗勒管化生或子宫内膜异位症的恶性转化。然而,包括本病例在内的许多已报道病例均缺乏子宫内膜异位症或激素影响的组织学证据。鉴于已报道的病例十分罕见,每一例新病例都能为这种未被充分认识的恶性肿瘤的临床和病理特征谱提供宝贵见解。本文报告一例无子宫内膜异位症或激素治疗史的 48 岁女性PPCCC病例,以凸显这种罕见肿瘤的诊断复杂性和临床意义。
病 例
患者女,48 岁,无明显既往病史或家族史,因急性下腹痛前往当地诊所就诊。她既往无激素替代治疗史或子宫内膜异位症病史。实验室检查显示肿瘤标志物升高,包括CA19-9(777.3 U/mL)和CA125(242.9 U/mL),而CEA(1.3 ng/mL)和AFP(3.06 ng/mL)处于正常范围内。腹部计算机断层扫描(CT)显示胰尾部有一个 15×10 mm的卵圆形、轻度强化病灶,怀疑为胰腺腺癌或胰岛细胞瘤。此外,左盆腔内可见一个 70×65×35 mm的边界清晰、异质性强化的实性肿块,怀疑为盆腔恶性肿瘤或转移灶。患者被转诊至三级医疗中心接受进一步评估。转诊后,盆腔磁共振成像(MRI)显示左侧附件区有一个 11 cm的囊实性混合肿块,实性成分呈异质性强化且弥散受限,提示为卵巢恶性肿瘤(图1)。临床初步诊断为卵巢和胰腺同时发生恶性肿瘤,患者接受了经腹全子宫切除加双侧输卵管卵巢切除术,以及远端胰腺脾切除术。术中发现盆腔腹膜内有一个巨大的盆腔肿块。术中冷冻切片分析初步提示为卵巢来源的高级别浆液性癌。胰尾部还发现一个 2 cm的包膜完整、边界清晰的肿块,附着于邻近的大网膜,需要进行粘连松解。但双侧卵巢、输卵管和子宫大体外观均无明显异常。
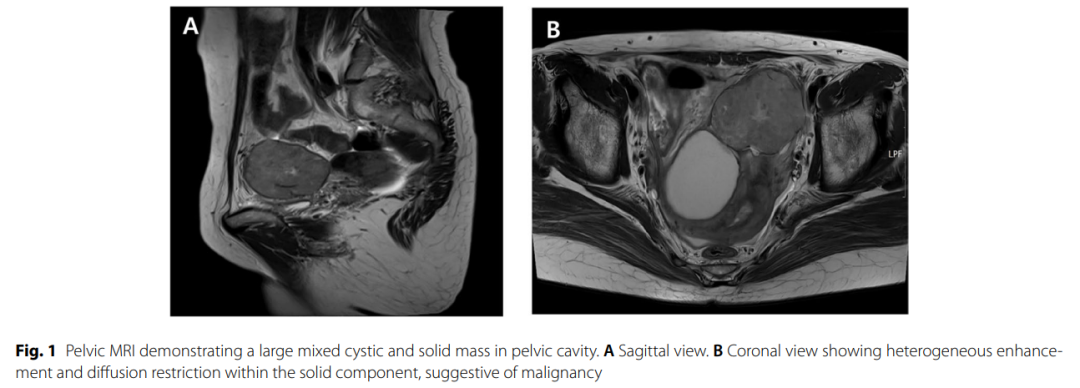
▲图1 盆腔MRI显示盆腔内有一个较大的混合性囊实性肿块
大体病理检查显示盆腔肿块大小为 12×11×3.5 cm,其他妇科器官未见明显病变(图2A-C)。为排除妇科来源,按照“输卵管伞端切开并广泛检查(SEE-FIM)”方案,将双侧卵巢和输卵管全部取材进行镜下检查,但未发现原发性肿瘤的证据。对盆腔肿块进行广泛取材,共提交约 30 个石蜡包埋组织块用于组织学评估。胰尾部可见一个 3×2.8×2.5 cm的囊性病变,腔内有易碎的白色肿块。脾实质内也发现了一个类似的 1.3×1.1 cm的囊性、易碎白色肿块样病变。镜下观察,胰腺病变以胰腺实质为中心,呈浸润性生长模式。肿瘤细胞形成实性和片状结构,伴广泛坏死,可见肿瘤细胞穿插于胰腺间质中,包绕并部分取代正常胰腺腺泡和导管。脾脏病变显示脾实质内存在囊性变,内含一个肿块状乳头状肿瘤。相邻红髓内可见肿瘤巢和腺管结构。所有部位的肿瘤均具有完全相同的组织形态学特征:肿瘤细胞核呈圆形至卵圆形,轮廓不规则,细胞质呈透明至嗜酸性,存在中度核异型性。核分裂活性不一,偶可见嗜酸性透明小体(图3A、B)。未发现淋巴血管浸润或神经周浸润。这些发现共同支持胰腺和脾脏病变均为实质真性受累。
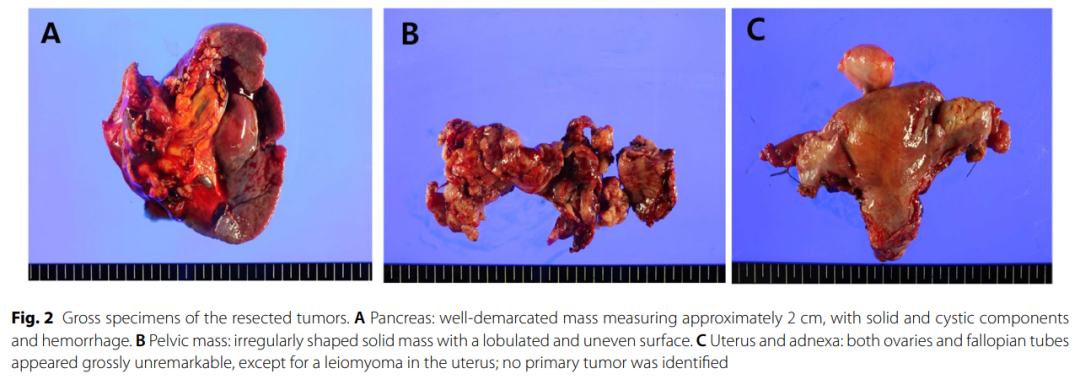
▲图2 切除肿瘤的大体标本
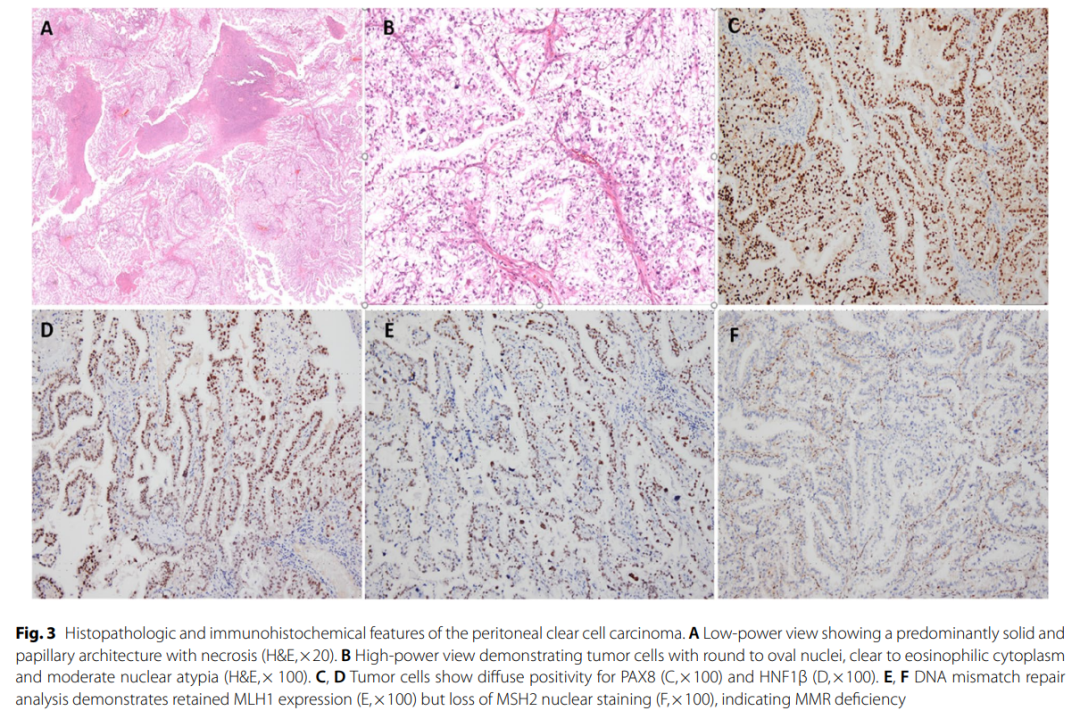
▲图3 腹膜透明细胞癌的组织病理学和免疫组织化学特征
标本也缺乏任何提示子宫内膜异位症的组织学特征,如子宫内膜腺体、间质或含铁血黄素巨噬细胞。免疫组化染色显示PAX8、CK7和HNF1β呈阳性,CK20、p16、WT1和钙视网膜蛋白呈阴性(图3C、D)。p53染色显示为野生型表达模式。PD-L1免疫组化染色在肿瘤细胞中呈阴性。DNA错配修复(MMR)免疫染色显示MSH2和MSH6的核表达缺失,而MLH1和PMS2表达保持完整,提示存在错配修复缺陷(图3E、F)。为排除透明细胞癌的妇科来源,对卵巢、输卵管和子宫进行了连续且全面的切片镜检,但未发现原发性肿瘤的证据。对肿瘤进行的下一代测序(NGS)显示存在ARID1A缺失,以及PIK3CA H1047R和PIK3CA Q546L的额外体细胞突变。基于术中发现、组织病理学特征、免疫组化表型及NGS检测的分子特征,最终诊断为原发性腹膜透明细胞癌伴胰腺和脾脏转移,这是一种极为罕见的临床表现。患者被诊断为IVB期原发性腹膜透明细胞癌,该病例已在多学科肿瘤委员会上讨论,以确定合适的辅助治疗方案。患者计划接受 6 个周期的吉西他滨联合卡铂化疗。
讨 论
原发性腹膜癌(PPC)是一种罕见的上皮恶性肿瘤,其特征为病变以腹膜为主,卵巢受累轻微或无受累。此前,妇科肿瘤学组(GOG)提出了区分PPC与原发性卵巢癌的操作标准,包括卵巢大小正常或仅为良性增大,且腹膜受累程度超过卵巢表面病变程度。然而,随着对盆腔高级别浆液性癌认知的进展,许多此前被归类为原发性腹膜浆液性癌的肿瘤,很可能起源于输卵管远端,因此“原发性腹膜来源”的传统概念已被大幅修正。与之不同的是,原发性腹膜透明细胞癌(PPCCC)的组织发生机制仍不明确,且尚未发现与浆液性输卵管上皮内癌类似的确定性输卵管前体病变。
PPCCC是PPC中极为罕见的组织学亚型,约占PPC病例的 3%。在组织学上,它与苗勒管来源的透明细胞癌(CCC)无法区分,尤其是卵巢和子宫内膜来源的透明细胞癌。因此,细致排除其他原发部位是确立该诊断的必要条件。在本病例中,对子宫、卵巢和输卵管进行的连续切片及详细组织病理学评估,均未发现原发性肿瘤的证据。尽管曾考虑过原发性透明细胞癌起源于输卵管的可能性,但采用SEE-FIM方案对双侧卵巢和输卵管进行的全面检查,未发现任何前体病变或浸润性病变,支持其为原发性腹膜来源。
虽然PPCCC的分子和发病机制尚未完全阐明,但最被广泛接受的假说认为其要么起源于苗勒管化生,要么来自子宫内膜异位症的恶性转化。不过,子宫内膜异位症的卵巢外恶性转化极为罕见,仅发生于 1.6% 的病例中。既往报道中,约三分之一的PPCCC患者有子宫内膜异位症病史,且组织学检查常能发现子宫内膜异位灶。但在本病例中,广泛的组织学取材未显示任何子宫内膜异位症的特征,如子宫内膜样腺体或间质,或含铁血黄素巨噬细胞。
表1汇总了对既往报道的PPCCC病例的综述,概述了患者人口统计学特征、肿瘤位置、子宫内膜异位症病史、分子检测结果及临床结局。大多数病例的发病年龄为 40-60 岁,仅部分病例存在子宫内膜异位症,反映了该恶性肿瘤发病机制的异质性。本病例具有与上述病例相似的组织病理学特征,但区别在于存在DNA错配修复缺陷,且NGS检测发现ARID1A缺失,凸显了二者分子特征的重叠性与独特性。腹膜来源CCC的鉴别诊断包括高级别浆液性癌、恶性间皮瘤,以及其他器官来源的转移性透明细胞癌,尤其是肾脏来源的。高级别浆液性癌的特征是核异型性显著、核分裂象多见,以及复杂的乳头状或实性结构。与之不同,恶性间皮瘤通常表现为相对温和的细胞学特征,以及由单层一致的间皮细胞排列形成的简单乳头状结构。尽管这些疾病与CCC存在形态学重叠,但通常可结合组织学评估和免疫组化表型进行区分。在转移性肿瘤中,透明细胞肾细胞癌(CCRCC)是重要的鉴别考量。CCRCC通常呈巢状-腺泡状生长模式,间质血管丰富,而苗勒管型CCC的特征是小管囊性结构、小圆形乳头和鞋钉样细胞。免疫组化方面,CCRCC通常CK7、HNF1β呈阴性,而妇科来源的CCC通常这些标志物呈阳性。在本病例中,肿瘤显示CK7和PAX8弥漫阳性,同时HNF-1β呈阳性,WT1呈阴性。DNA错配修复(MMR)分析显示MSH2和MSH6的核表达缺失,而MLH1和PMS2表达保持完整,符合错配修复缺陷的特征。p53染色显示为野生型模式。这些发现共同支持腹膜来源的妇科透明细胞癌的诊断。
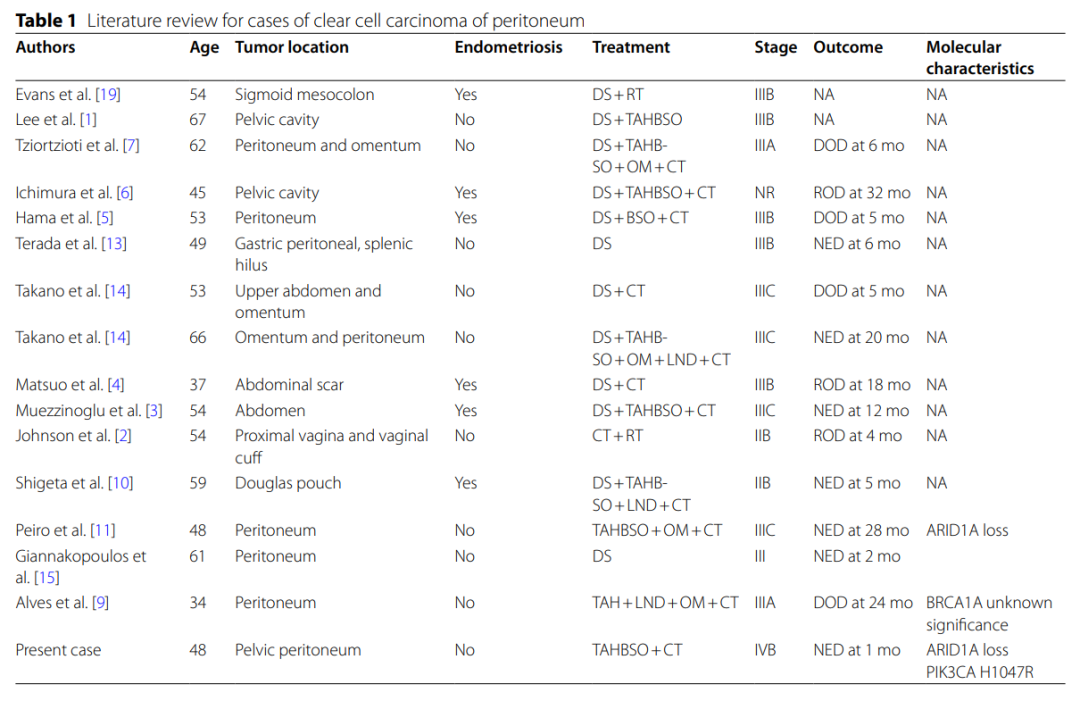
▲表1 腹膜透明细胞癌病例的文献综述
错配修复(MMR)缺陷的检出具有重要的临床意义。MSH2和MSH6表达缺失提示可能存在潜在的林奇综合征,因此需要进一步进行遗传咨询和胚系检测。此外,错配修复缺陷与肿瘤突变负荷升高相关,即使是在透明细胞癌这类通常被认为具有化疗耐药性的恶性肿瘤中,该特征也可能提示肿瘤对免疫检查点抑制剂具有潜在应答性。
近期的分子研究揭示了卵巢透明细胞癌相关的基因改变,报道显示ARID1A、PIK3CA和KRAS突变在该疾病中频发。Peiró等人也在一例PPCCC病例中描述了ARID1A的致病性缺失,以及GSDMB和KMT2C的额外体细胞突变。在本病例中,分子分析显示存在ARID1A缺失,与Peiró等人报道的结果一致。此外,下一代测序检测到PIK3CA H1047R突变,进一步丰富了该患者的PPCCC分子谱。PIK3CA是PI3激酶的催化亚基,在乳腺癌、子宫内膜癌和宫颈癌等多种癌症中频发突变。已知H1047R突变具有致癌性。临床前数据显示,携带这类PIK3CA突变的肿瘤可能对RLY-2608等同工酶选择性PIK3CA抑制剂敏感,这为潜在的靶向治疗方案提供了思路。尽管目前关于PPCCC的数据仍然有限,但这些重叠的分子特征提示其可能与卵巢透明细胞癌具有共同的发病机制,凸显了进一步开展分子研究以更好地描述这种罕见恶性肿瘤特征的重要性。此外,潜在可治疗突变的检出,也凸显了全面分子检测在指导未来研究和个体化治疗策略中的作用。
临床表现方面,PPCCC通常表现为腹痛、腹水和盆腔肿块,本病例患者即符合上述表现。虽然大多数已报道的病例病变仍局限于腹膜腔内,但也有文献记录了皮肤、脑等腹膜腔外的远处转移。本病例中胰腺和脾脏的转移受累提示疾病已处于进展期(FIGO IVB期),印证了PPCCC常伴随的侵袭性临床行为和不良预后。由于PPCCC的罕见性,目前尚无成熟的治疗策略,通常参考上皮性卵巢癌的治疗方案,包括肿瘤细胞减灭术术后序贯铂类为基础的化疗。即便接受这类治疗,患者的预后仍不理想,常出现早期复发,且对标准方案应答有限。靶向PD-1/PD-L1的免疫检查点抑制剂已在透明细胞癌亚型中进行了探索,但其疗效有限。尽管本病例的PD-L1表达为阴性,但错配修复缺陷的存在仍可为疾病进展或复发时考虑免疫治疗提供依据。
综上,PPCCC是一种罕见的侵袭性恶性肿瘤,给诊断和治疗带来了巨大挑战。准确诊断需要全面的组织学、免疫组化和临床评估,以排除更常见的原发部位肿瘤。本病例为有限的相关文献提供了新的补充,凸显了开展进一步研究以探究这种未被充分认识的恶性肿瘤的发病机制、分子特征和潜在靶向治疗方案的必要性。
结 论
原发性腹膜透明细胞癌(PPCCC)是一种极为罕见的恶性肿瘤,由于其罕见性,且与该部位更常见的肿瘤特征存在重叠,因此诊断仍具有挑战性。在很多病例中,它可能未被充分识别,或被误诊为卵巢或其他器官来源的转移性疾病。因此,全面排除其他潜在原发部位至关重要。除了特征性的组织病理学特征外,免疫组化在鉴别诊断中也发挥着关键作用,有助于将PPCCC与卵巢、肾脏和间皮来源的肿瘤区分开。准确识别这种罕见恶性肿瘤,对于实现精准诊断、指导制定适宜的治疗策略至关重要。鉴于PPCCC具有侵袭性行为且预后不佳,需要开展进一步研究以更好地了解其分子发病机制,并建立优化的治疗方案,从而降低死亡率。
参考文献:
Kim, N.I., Lee, J.S., Lee, K.H. et al. Primary peritoneal clear cell carcinoma with metastasis mimicking ovarian carcinoma: a case report and literature review. J Ovarian Res 19, 111 (2026). https://doi.org/10.1186/s13048-026-02009-w

