分子检测揭示灰色地带:1p/19q共缺失但IDH野生型的高级别胶质瘤,如何精准分类?
时间:2026-06-16 20:12:29 热度:37.1℃ 作者:网络
深度解析医学证据,lxfs.net为你支撑决策
胶质瘤是最常见的颅内恶性肿瘤,预后不良。2016 年世界卫生组织(WHO)整合分类将弥漫性胶质瘤主要基于异柠檬酸脱氢酶(IDH)基因突变状态和1p/19q共缺失状态进行分类,将弥漫性胶质瘤分为三个不同的分子类别:染色体1p/19q共缺失/IDH突变型、1p/19q完整/IDH突变型和IDH野生型。携带1p/19q共缺失但无IDH突变的胶质瘤较为罕见,且无法根据WHO新修订的分类标准进行分类。本文报告了三例具有这种非典型分子表型的高级别胶质瘤,并描述了组织学和免疫组织化学特征、TERT启动子、H3F3A、HIST1H3B和BRAF基因的突变状态、MGMT启动子甲基化状态以及预后情况。综合考虑形态学、分子参数和患者预后,发现携带1p/19q共缺失但无IDH突变的高级别胶质瘤并非典型的胶质母细胞瘤(GBM),但相较于间变性少突胶质细胞瘤,它们更倾向于GBM。
背 景
胶质瘤是最常见的颅内恶性肿瘤,预后不良。I级和II级胶质瘤为低级别,而III级和IV级胶质瘤为高级别,后两者预后更差。以往,胶质瘤的诊断完全基于形态学。近年来分子分析技术的发展揭示了胶质瘤中许多遗传学改变。2016 年世界卫生组织(WHO)中枢神经系统(CNS)肿瘤分类将分子信息整合到CNS肿瘤分类中,打破了百年来完全基于组织学诊断的原则。这种新的弥漫性胶质瘤整合分类主要基于异柠檬酸脱氢酶(IDH)基因突变状态和1p/19q共缺失状态,将弥漫性胶质瘤分为三个不同的分子类别:染色体1p/19q共缺失/IDH突变型、1p/19q完整/IDH突变型和IDH野生型。在新分类中,少突胶质细胞瘤被定义为具有IDH1和IDH2突变以及1p/19q共缺失的弥漫浸润性胶质瘤。
其他分子生物标志物,包括端粒酶逆转录酶(TERT)基因启动子突变、H3K27突变、BRAF突变和MGMT启动子甲基化,已广泛用于胶质瘤的诊断、治疗和预后预测。TERT启动子突变和MGMT启动子甲基化也存在于绝大多数少突胶质细胞瘤中。α-地中海贫血/智力低下综合征X连锁(ATRX)蛋白表达的缺失与1p/19q共缺失互斥。强烈且弥漫的核p53染色常见于胶质母细胞瘤(GBM)。
1p/19q共缺失是少突胶质细胞瘤最重要的遗传学特征。根据一些研究,典型的1p/19q共缺失总是与IDH突变相关。然而,也有携带1p/19q共缺失但无IDH突变的胶质瘤的报道,其发生率约为 0.2%-0.4%。这些特殊病例均未进行过多基因综合分子检测(如TERT启动子突变和MGMT启动子甲基化),也未在既往研究中得到分析。因此,对这些胶质瘤进行分类较为困难。
本研究报告了三例表现为IDH野生型和1p/19q共缺失的高级别胶质瘤病例。研究人员全面描述了它们的临床和组织学特征、免疫组织化学表型以及其他分子特征,如TERT启动子、H3F3A、HIST1H3B和BRAF的突变状态以及MGMT启动子甲基化状态,并讨论了此类灰色地带胶质瘤的合适分类。
病 例
病例1
患者女,38 岁,因反复头痛三年就诊,一个月前有癫痫发作史。进行性头痛伴非喷射性呕吐 15 天。磁共振成像(MRI)显示右侧额叶T1加权像(T1WI)和T2加权像(T2WI)上见高信号肿块(图1A、B)。可见少量斑片状水肿区,在T1WI上呈低信号,在液体衰减反转恢复(FLAIR)像上呈高信号(图1C)。钆增强T1WI显示肿瘤周边明显强化(图1D)。手术切除肿块。术后患者拒绝辅助放疗和化疗,术后 9 个月死亡。
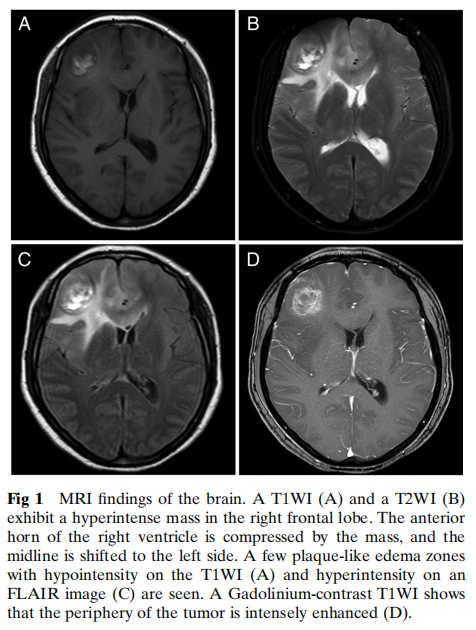
图1 脑部MRI检查结果
病例2
患者男,63 岁,因进行性头痛十天入院。MRI显示右侧颞叶和顶叶有一处病变,考虑为高级别胶质瘤。术后接受了放疗,并辅以长期替莫唑胺(TMZ)辅助治疗。然而,六个月后肿瘤复发并进展,出现颅内多发转移。术后十二个月死亡。
病例3
患者男,48 岁,因右手麻木且只能缓慢书写转入我院。MRI显示左侧顶叶有一边界不清的肿块,伴不均匀强化区域。患者接受了肿块全切除术及标准替莫唑胺化疗。截至本文撰写时,术后仅过去两个月,未见局部复发或远处转移征象。
研究材料和方法
研究材料
采用存档的福尔马林固定、石蜡包埋(FFPE)标本进行组织病理学、免疫组织化学和分子分析。所有材料的使用均遵循机构批准的伦理指南和程序。
免疫组化
免疫组织化学方面,使用了针对以下指标的一抗:IDH1 R132H(小鼠单克隆抗体,克隆号H09)、p53(小鼠单克隆抗体,克隆号DO-7)、Ki-67(小鼠单克隆抗体,克隆号MIB-1)、胶质纤维酸性蛋白(GFAP)(兔单克隆抗体,克隆号EP13)、Oligo2(兔单克隆抗体,克隆号EP112)、ATRX(兔多克隆抗体,ZA-0016)、上皮膜抗原(EMA)(小鼠单克隆抗体,克隆号E29)、NeuN(小鼠单克隆抗体A60)以及H3 K27M(兔单克隆抗体,克隆号RM192)。免疫反应产物沉积采用Envision系统的聚合物免疫复合物法进行可视化,操作按照制造商的方案进行。组织学观察在苏木精和伊红(HE)染色切片上进行。
荧光原位杂交
荧光原位杂交(FISH)使用双色1p36/1q25和19q13/19p13探针进行。简而言之,将FFPE切片脱蜡、透化并进行杂交。两个探针组在超过 90% 的细胞核中均显示明亮信号的样本视为合格。在荧光显微镜下对 100 个非重叠细胞核进行1p36(红色)/1q25(绿色)和19q13(红色)/19p13(绿色)探针信号的计数。当 30% 或以上的细胞核仅显示一个红色信号和两个绿色信号时,判定为缺失。
基于下一代测序技术的靶向单核苷酸多态性分析
下一代测序(NGS)按照制造商的方案操作。根据文献,通过NGS检测染色体1p和19q的单核苷酸多态性(SNP)。分别选取了1p染色体上的 28 个靶标SNP和19q染色体上的 32 个靶标SNP。当检测正常样本时,B等位基因频率在 35% 至 65% 之间变化,纯合参考和纯合变异分别位于接近 0% 和 100% 的等位基因频率处。当检测存在1p或19q缺失的样本时,B等位基因频率发生偏移。
基于聚合酶链反应的Sanger测序
使用试剂盒按照制造商说明提取总DNA。聚合酶链反应(PCR)引物设计如下:IDH1(上游引物,5'-CGG TCT TCA GAG AAG CCA TT-3';下游引物,5'-GCA AAA TCA CAT TAT TGC CAA C-3',129 bp)、IDH2(上游引物,5'-AGC CCA TCA TCT GCA AAA AC-3';下游引物,5'-CTA GGC GAG GAG CTC CAG T-3',150 bp)、H3F3A(上游引物,5'-GTA CAA AGC AGA CTG CCC GCA AAT-3';下游引物,5'-GTG GAT ACA TAC AAG AGA GAC TTT GTC CC-3',173 bp)、HIST1H3B(上游引物,5'-CTG CTC GTA AGT CCA CCG GTG-3';下游引物,5'-GCG ATC TCC CTC ACC AAC CTC-3',243 bp)、TERT启动子(上游引物1,5'-GTC CTG CCC CTT CAC CTT-3';下游引物1,5'-GCA CCT CGC GGT AGT GG-3',273 bp;上游引物2,5'-CCG TCC TGC CCC TTC ACC-3';下游引物2,5'-GGG CCG CGG AAA GGA AG-3',128 bp)以及BRAF(上游引物,5'-TCA TAA TGC TTG CTC TGA TAG GA-3';下游引物,5'-GGC CAA AAA TTT AAT CAG TGG A-3',223 bp)。使用标准缓冲液条件、50 ng DNA和Taq DNA聚合酶。反应混合物先在 95°C 下初始变性 10 分钟,然后进行 35 个循环的扩增(95°C 变性 30 秒,56°C 退火 30 秒,72°C 延伸 40 秒)。PCR产物使用MinElute PCR纯化试剂盒进行纯化,并使用自动测序仪进行正义链和反义链双向测序。
MGMT启动子甲基化
从FFPE切片中提取总DNA用于亚硫酸氢盐转化。通过甲基化特异性PCR评估MGMT甲基化状态,该PCR使用特异性针对甲基化和非甲基化MGMT启动子序列的PCR引物组:MGMT甲基化引物,5'-TTT CGA CGT TCG TAG GTT TTC GC-3',5'-GCA CTC TTC CGA AAA CGA AAC G-3';MGMT非甲基化引物,5'-TTT GTG TTT TGA TGT TTG TAG GTT TTT GT-3',5'-AAC TCC ACA CTC TTC CAA AAA CAA AAC A-3'。
研究结果
神经病理学检查结果
所有三例病例的组织学检查均显示高度细胞和细胞核多形性,出现大量多核巨细胞及活跃的核分裂象。所有病例均可见明显的微血管增生,病例3可见栅栏状坏死,但病例1和2未见。病例2和3中还观察到少突胶质细胞瘤样成分,但病例1未见。在病例2中,肿瘤细胞中散在分布着一种特定的神经毡样岛状结构。代表性形态学见图2、图3和图4。
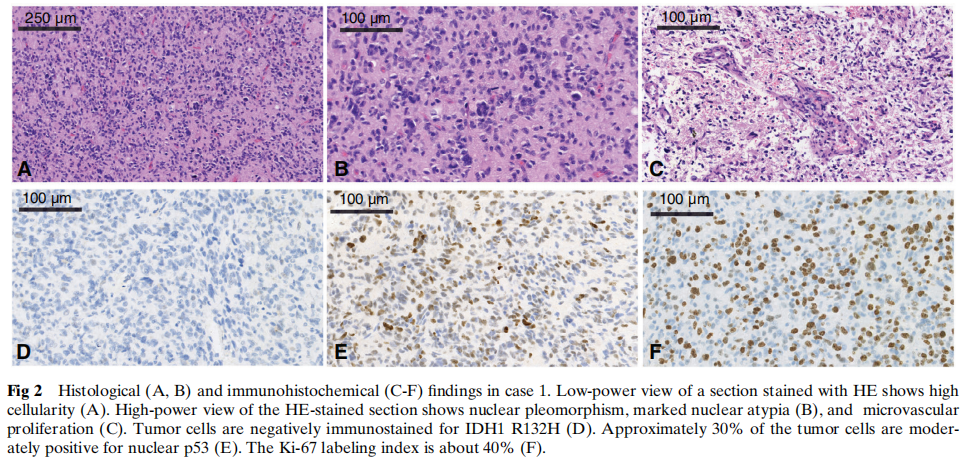
图2 病例1的组织学(A、B)及免疫组织化学(C–F)检查结果
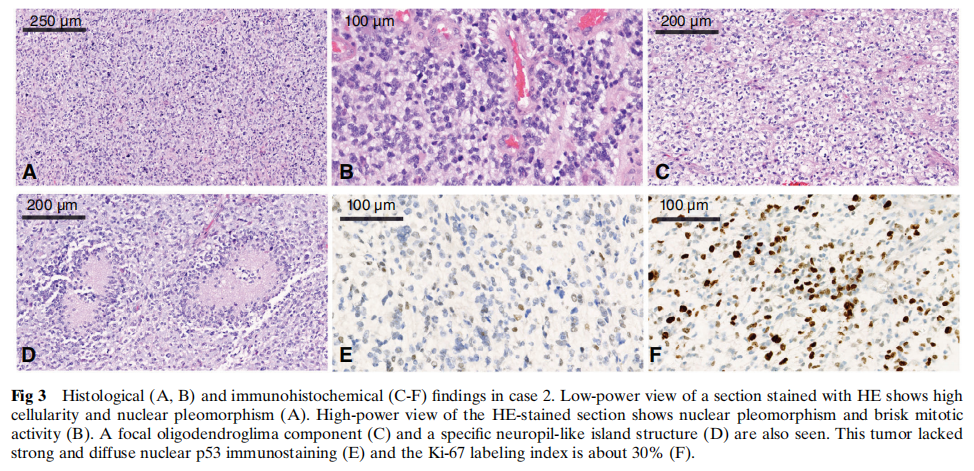
图3 病例2的组织学(A、B)及免疫组织化学(C–F)检查结果
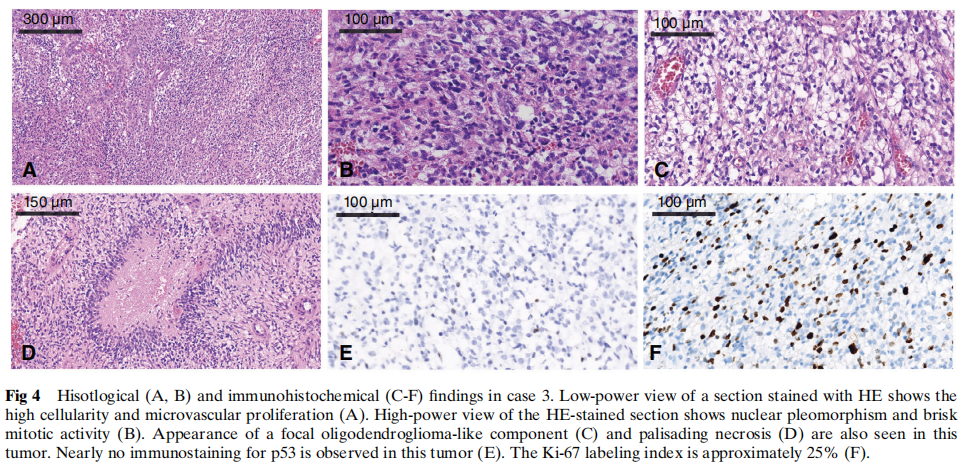
图4 病例3的组织学(A、B)及免疫组织化学(C-F)检查结果
免疫组织化学方面,所有病例的肿瘤细胞均呈GFAP和Oligo2强阳性。ATRX在细胞核中保留表达。IDH1 R132H、EMA、NeuN和H3 K27M在所有病例中均为阴性。根据Ki-67标记指数(25%-40%),肿瘤细胞具有高增殖活性,且在不同病例中,约 5% 至 30% 的肿瘤细胞呈p53弱阳性。病例1、2和3的分子改变免疫组织化学结果汇总于表1及图2、图3和图4。
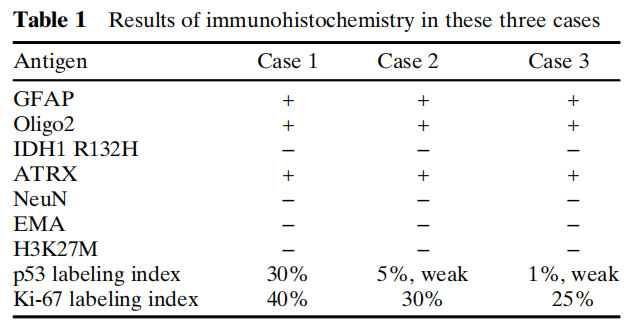
表1 三例病例的免疫组织化学检测结果
通过FISH分析(图5A)和基于NGS的SNP分析(图5B),三例病例均证实存在1p/19q共缺失。其他分子分析中未检测到热点突变,包括IDH1(R132)、IDH2(R172)、H3F3A(K27和G34)、HIST1H3B(K27和G34)、BRAF(V600)和TERT启动子(228和250)。基于甲基化特异性PCR,病例2中检测到MGMT启动子甲基化,而病例1和3中未检测到。遗传学改变汇总于表2。
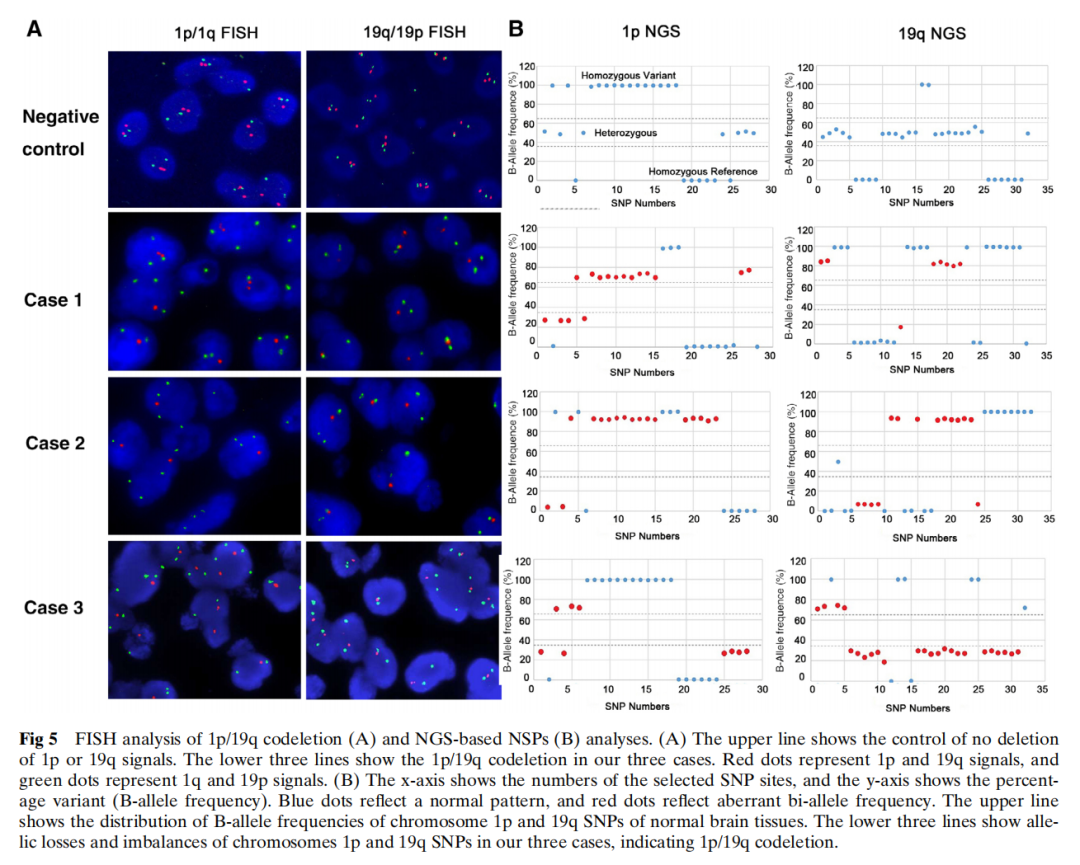
图5 1p/19q共缺失的FISH分析(A)及基于NGS的非编码序列分析(B)
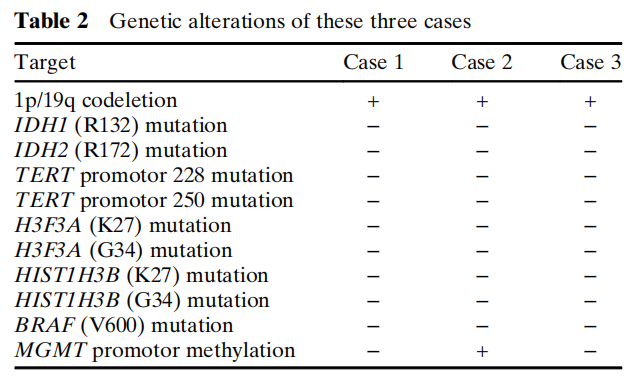
表2 三例病例的基因变异
讨 论
2016 年WHO中枢神经系统肿瘤分类利用分子遗传学数据来区分不同类型的胶质瘤,打破了传统的分类方式。人们希望新分类能够利用分子遗传学标准(而非以往仅基于形态学的分级标准)更清晰地提示肿瘤行为。根据新分类,少突胶质细胞瘤仅在同时携带IDH1或IDH2突变和1p/19q共缺失时才能被诊断。如果通过免疫组织化学和DNA测序均证实IDH为野生型,则不推荐进行1p/19q FISH检测。因此,发现IDH野生型且伴有1p/19q共缺失的胶质瘤并不容易,迄今为止相关研究也很少。这类胶质瘤是独立的实体还是属于某种特定的胶质瘤亚型,目前尚不清楚。
WHO分类并未建议使用哪种方法来检测1p/19q共缺失,但指出应检测整条染色体臂的缺失。目前评估1p/19q的方法包括FISH、显色原位杂交(CISH)、基于PCR的微卫星分析、阵列比较基因组杂交(CGH)、多重连接依赖性探针扩增(MLPA)、SNP阵列和NGS。FISH是评估1p和19q缺失最常用的方法之一,尤其是在医院。FISH的优点是能够保留组织结构和细胞学特征,且评估基于形态学。其缺点是探针无法覆盖整条染色体臂,且无法区分整臂缺失和部分缺失。相比之下,其他方法如基于PCR的微卫星分析、CGH、MLPA、SNP阵列和NGS可以评估1p和19q的整臂缺失,但无法进行原位检测。为确保结果的准确性,研究人员同时使用了FISH分析和基于NGS的SNP分析来评估1p和19q染色体的状态,并确认了本研究中病例的1p/19q共缺失。
尽管大多数研究表明典型的1p/19q缺失总是与IDH突变相关,但至少已有八例伴有1p/19q共缺失和IDH野生型的胶质瘤被报道。根据两项近期根据 2016 年新分类对弥漫性胶质瘤进行重新分类的大规模分子遗传学研究,在 1041 例胶质瘤中仅有 4 例,在 1206 例胶质瘤中仅有 2 例检出具有这些非典型分子改变。然而,由于非典型分子表型,这些病例在重新分配时被排除,未进行进一步的全面分子分析。在另一项对 82 例胶质瘤进行重新分类的研究中,两例伴有1p/19q共缺失和IDH野生型的肿瘤被重新归类为少突胶质细胞瘤,NOS。
本研究报告了三例基于免疫组织化学和DNA测序结果均显示伴有1p/19q共缺失但无IDH1/2热点突变的高级别胶质瘤;根据修订后的WHO分类,这些属于灰色地带病例。众所周知,高细胞密度、细胞多形性、显著的微血管增生和/或栅栏状坏死是GBM的基本诊断特征。在本研究中,所有病例均可见高度细胞和细胞核多形性及微血管增生,但典型的栅栏状坏死仅见于病例3。病例2和3中可见部分少突胶质细胞瘤样区域,根据 2007 年形态学标准,这导致诊断为间变性少突星形细胞瘤或伴有少突胶质细胞瘤成分的胶质母细胞瘤(GBMO)。随后对这三例病例全面检测了其他胶质瘤相关遗传指标。在这些病例中,ATRX表达保留,p53标记指数非常低,且p53免疫反应性相对较弱。所有病例均未检测到H3F3A、HIST1H3B、BRAF和TERT启动子的热点突变。病例2中确认存在MGMT启动子甲基化,病例1中未检测到甲基化。病例1和2的预后相对较差,与GBM相似。综合考虑这些病理学、分子特征及生物学行为,研究人员得出结论:这些罕见肿瘤并非非典型GBM,但相较于间变性少突胶质细胞瘤,它们更接近GBM。
总之,本文报告了一种具有罕见分子表型的特定类型高级别胶质瘤,基于免疫组织化学和DNA测序结果,该肿瘤伴有1p/19q共缺失但无IDH1/2热点突变。根据 2016 年修订版WHO中枢神经系统肿瘤分类的分子遗传学标准,这些胶质瘤无法进行分类。综合考虑形态学、其他分子指标以及患者预后,研究人员发现这些胶质瘤更倾向于GBM而非间变性少突胶质细胞瘤。然而,需要更多病例来对该罕见肿瘤进行准确分类。
参考文献:
Zheng L, Zhang M, Hou J, Gong J, Nie L, Chen X, Zhou Q, Chen N. High-grade gliomas with isocitrate dehydrogenase wild-type and 1p/19q codeleted: Atypical molecular phenotype and current challenges in molecular diagnosis. Neuropathology. 2020 Dec;40(6):599-605. doi: 10.1111/neup.12672. Epub 2020 Aug 5. PMID: 32761642.

